


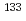 CsI (v = 0, j = 0) +
CsI (v = 0, j = 0) + 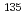 Cs
Cs  Cs + ICsCsI (v=0, j=0) + Cs Cs + ICsの準古典的トラジェクトリー計算
Cs + ICsCsI (v=0, j=0) + Cs Cs + ICsの準古典的トラジェクトリー計算Kobayashi, Takanori*; Matsuoka, Leo*; Yokoyama, Keiichi
セシウム交換反応CsI (v=0, j=0) + Cs Cs + ICsの反応断面積を調べるため、ab initio分子軌道法計算により作成したポテンシャルエネルギー面を用いた準古典的トラジェクトリー計算を行った。ポテンシャルエネルギー面から反応中間体Cs Iの生成に入口障壁がないこと、2つのCsI結合が等価であることが明らかになった。トラジェクトリー計算により反応断面積は衝突エネルギーの増加と共に単調増加することが分かった。CsI分子の初期内部状態がv=0, j=0の場合の反応速度定数は500-1200Kの温度範囲で3
Iの生成に入口障壁がないこと、2つのCsI結合が等価であることが明らかになった。トラジェクトリー計算により反応断面積は衝突エネルギーの増加と共に単調増加することが分かった。CsI分子の初期内部状態がv=0, j=0の場合の反応速度定数は500-1200Kの温度範囲で3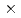 10
10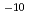 cm
cm molecule
molecule s程度と見積もられ、わずかながら負の温度依存性が見られた。
s程度と見積もられ、わずかながら負の温度依存性が見られた。
To investigate the reaction cross section of the cesium exchange reaction of CsI (v = 0, j = 0) + Cs Cs + ICs, we performed quasi-classical trajectory calculations on the potential energy surface calculated by the ab initio molecular orbital theory. The potential energy surface shows that intermediate CsI is formed without entrance barrier and has two equivalent Cs-I bonds. The reaction cross sections decrease monotonically with increasing collision energy. The rate constant k (v = 0, j = 0) was estimated to be about 310cm molecules at temperatures ranging from 500 to 1200K and a slight negative temperature dependence was observed.
使用言語 |
: | English |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | 96 |
号 |
: | 10 |
ページ数 |
: | p.441 - 444 |
発行年月 |
: | 2017/10 |
キーワード |
: |
論文URL |
: |
|
|---|---|---|
使用施設 |
: | |
論文解説記事 |
: |
Access |
: |
- Accesses |
|---|---|---|
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.


