


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Xianglian*; 佐久間 隆*; Mohapatra, S. R.*; 上原 寛之*; 高橋 東之*; 神嶋 修*; 井川 直樹
Molecular Simulation, 38(5), p.448 - 451, 2012/04
被引用回数:4 パーセンタイル:9.26(Chemistry, Physical)10K及び294KにおけるPbTeの中性子散漫散乱実験を行った結果、PbTeの散乱強度の振動項は原子の熱変位における相関効果によって説明できることを明らかにした。相関効果とDebye-Waller温度因子から計算した294Kにおける第1近接原子間の力定数は
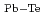 =215eV/nm
=215eV/nm と、第2近接原子間のそれは、各々
と、第2近接原子間のそれは、各々 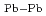 =108eV/nm及び
=108eV/nm及び 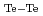 =144eV/nmと求めることができた。
=144eV/nmと求めることができた。
 quantum mechanical/molecular mechanical molecular dynamics using multiple-time-scale approach and perturbation theory
quantum mechanical/molecular mechanical molecular dynamics using multiple-time-scale approach and perturbation theory志賀 基之; 立川 仁典*
Molecular Simulation, 33(1-2), p.174 - 184, 2007/02
第一原理量子古典分子動力学法は法による非経験的な力場計算と経験的な力場計算を組合せたハイブリッド法で、凝集分子系に対して有効なシミュレーション手法として最近注目を集めている。本研究では、第一原理量子古典分子動力学法を用いて熱力学的解析を行うのに適した手法を提案し、その計算効率や計算精度について議論する。また、この手法の有用性を確かめるため、水溶液の構造とエネルギーの計算結果を報告する。
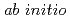 path integral molecular dynamics simulation
path integral molecular dynamics simulation林 愛子*; 志賀 基之; 立川 仁典*
Molecular Simulation, 33(1-2), p.185 - 188, 2007/02
被引用回数:3 パーセンタイル:9.14(Chemistry, Physical)近年、二水素結合やリチウム-水素結合など新しいタイプの水素結合様式が実験的に見いだされているが、これらの水素結合の性質や構造は詳細にわかっていないものも多い。これらに対し、第一原理経路積分分子動力学法を用いてC H
H LiH分子クラスターの計算機シミュレーションを行った結果、これらの結合においても通常の水素結合と同様に大きな同位体効果があることがわかった。
LiH分子クラスターの計算機シミュレーションを行った結果、これらの結合においても通常の水素結合と同様に大きな同位体効果があることがわかった。
米谷 佳晃*; 河野 秀俊; 藤井 聡*; 皿井 明倫*; 郷 信広
Molecular Simulation, 33(1-2), p.103 - 107, 2007/01
被引用回数:5 パーセンタイル:16.57(Chemistry, Physical)5'AATT3'と5'TTAA3'の2種類の塩基配列のDNAについて分子動力学シミュレーションを行い、構造変化と水和の関係を調べた。シミュレーションから、5'AATT3'では、DNAは構造変化しにくく、水和水は構造化しやすいが、5'TTAA3'では、DNAは構造変化しやすく、水和水は構造化しやすいことが明らかになった。この結果に基づいてDNAの構造変化と水和の関係について議論した。
神林 奨; 千原 順三
Molecular Simulation, 16, p.31 - 46, 1996/00
被引用回数:4 パーセンタイル:17.3(Chemistry, Physical)従来のカーパリネロの理論による分子動力学法とは異なった、新しい第1原理的分子動力学法(QHNC-MD法)を考案した。QHNC-MD法では、液体金属中の電子及びイオンに関する動径分布関数と有効イオン間ポテンシャルに対する量子的HNC方程式を、古典的分子動力学シミュレーションを用いて解く方法である。この方法では、分子動力学シミュレーションによって得られるイオン間分布関数と有効イオン間ポテンシャルを自己無撞着に決定することが可能である。また、QHNC-MD法による有効イオン間ポテンシャルの収束計算は高速であり、しかも、数千から数万個の規模のシミュレーションが可能である。この点はカーパリネロ手法と大きく異なる部分である。液体アルカリ金属に関するQHNC-MDシミュレーションから得られた静的構造因子は、X線・中性子線実験の結果と極めて良く一致し、従来のQHNC方程式の近似解に見られる欠点を取り除くことが可能となった。
蕪木 英雄; 横川 三津夫
Molecular Simulation, 12(3-6), p.441 - 444, 1994/00
被引用回数:3 パーセンタイル:27.45(Chemistry, Physical)希薄気体の数値シミュレーション手法のひとつである直接シミュレーション・モンテカルロ法(DSMC法)について、連続領域の流れへの適用可能性について検討した。クヌーセン数2.96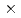 10
10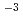 、レイノルズ数100の2次元キャビティ内の連続領域の流れに対し、DSMC法と有限差分法とを比較した結果、DSMC法は連続領域の流れの数値シミュレーションに、十分適用可能であることを見い出した。
、レイノルズ数100の2次元キャビティ内の連続領域の流れに対し、DSMC法と有限差分法とを比較した結果、DSMC法は連続領域の流れの数値シミュレーションに、十分適用可能であることを見い出した。
千原 順三; 石飛 昌光*
Molecular Simulation, 12(3-6), p.187 - 195, 1994/00
被引用回数:3 パーセンタイル:14.83(Chemistry, Physical)イオン電子混合系とみなせる液体金属においては、その分子動力学を行うとき、常に2体の原子間ポテンシャルで正確に記述できることを示した(多体力は不要)。
町田 昌彦*; 蕪木 英雄
Molecular Simulation, 12(3-6), p.435 - 439, 1994/00
被引用回数:0 パーセンタイル:0.15(Chemistry, Physical)絶縁体などの電子が関与しない系での熱伝導メカニズムを調べるため、我々は一次元非線形格子モデルのコンピュータシミュレーションを行った。用いたモデルは、2原子戸田格子とFPUモデルの2つであり、いづれも両端を固定した境界条件で、一定のエネルギーを粒子の運動量にランダムに付与する。この条件を下にニュートンの運動方程式を、鈴木・トロッター分解の4次精度の手法を用いて数値的に解く。得られる量は、時々刻々の座標と運動量であり、これらを用いて2つの初期条件をわずかにずらした系の起動の分離距離を求める。この量は、系の運動の不定性及びカオスの強さを知る指標的な量であり、モデルの違いによるカオスの度合いの違いを知ることができる。これによりカオスの度合いは、非線形性を上げることで強くなり、これは、上記2つのモデルにおける熱伝導シミュレーションの結果を説明できることがわかった。
神林 奨; 樋渡 保秋*
Molecular Simulation, 12(3-6), p.421 - 430, 1994/00
被引用回数:3 パーセンタイル:14.83(Chemistry, Physical)逆ベキポテンシァルによって粒子間相互作用が記述されるソフト球モデルについて、粒子数13,500個の等温分子動力学シミュレーションを実行した。このシミュレーションによって得られた2体分布関数を用いて、液体の分布関数理論において重要なブリッジ関数を数値的に計算した。計算結果とRogers-Young(RY)近似およびMHNCS近似とを比較したところ、RY近似では、過冷却領域における振動的なブリッジ関数の特徴を再現できないが、MHNCS近似では、この特徴を非常によく再現していることが明らかになった。RY近似およびMHNCS近似の違いは、前者に基本ダイアグラムによって示される粒子相関が含まれておらず、後者に含まれている点である。シミュレーションから得られた結果は、過冷却状態において、基本ダイアグラムによって表される粒子相関がきわめて重要であることを示している。
