


 CHO
CHO CH+HCO; Direct ab initio molecular dynamics study
CH+HCO; Direct ab initio molecular dynamics studyKurosaki, Yuzuru; Yokoyama, Keiichi 

A total of 400 trajectories for the photodissociation, CHCHOCH+HCO, on the T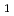 potential surface have been calculated using the direct ab initio molecular dynamics method at the UB3LYP/cc-pVDZ level of theory. It was predicted that the product CH is neither vibrationally nor rotationally excited and HCO is vibrationally not excited but rotationally excited. The averaged HCO rotational energy was calculated to be 1.1 kcal/mol, which is 15.1 % of the available energy, 7.3 kcal/mol. The present result agrees with experiment within just a few percent of the observed data.
potential surface have been calculated using the direct ab initio molecular dynamics method at the UB3LYP/cc-pVDZ level of theory. It was predicted that the product CH is neither vibrationally nor rotationally excited and HCO is vibrationally not excited but rotationally excited. The averaged HCO rotational energy was calculated to be 1.1 kcal/mol, which is 15.1 % of the available energy, 7.3 kcal/mol. The present result agrees with experiment within just a few percent of the observed data.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 371 |
Number |
: | 5-6 |
Pages |
: | p.568 - 575 |
Publication Year/Month |
: | 2003/04 |
Paper URL |
: |
|
Keywords |
: | Acetaldehyde; Photodissociation; Ab Initio Molecular Dinamics; Density Functional; Excited Triplet State |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:68.69 Category:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
