


 /SiC界面原子構造における界面接続モデル/SiC interface atomic structure generated with first-principles molecular dynamics simulation
/SiC界面原子構造における界面接続モデル/SiC interface atomic structure generated with first-principles molecular dynamics simulation宮下 敦巳; 大沼 敏治*; 岩沢 美佐子*; 土田 秀一*; 吉川 正人
Miyashita, Atsumi; Onuma, Toshiharu*; Iwasawa, Misako*; Tsuchida, Hidekazu*; Yoshikawa, Masahito
SiCを用いた半導体デバイスは、従来のSiやGaAsでは動作が困難な環境下でも使用可能なデバイスとして期待されているが、SiC MOS-FETでは、SiCと酸化膜の界面にデバイス特性を劣化させる界面欠陥が多く存在しているため、その欠陥構造とデバイス特性との関連性を追求することが重要な課題となっている。本研究では実際の界面を模擬した原子構造モデルを計算機上に生成し、その電子状態が界面電気特性に与える影響を理論的側面から追求している。4H-SiC(0001)上に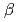 水晶を接続した界面原子構造モデルに対して加熱・急冷計算を行い、アモルファスSiO/SiC界面原子構造を生成した。なお、加熱・急冷計算には第一原理分子動力学計算コードであるVASPを用いた。生成された界面構造モデルでは、従来モデルにおいて想定されていた、Si原子が界面にある2つないしは3つのSi-O結合をまとめる界面接続モデル以外にも、Si原子が一つのSi-O結合にのみ接続する構造ができていた。加えて従来モデルでは想定されていなかった、Siダングリングボンド,Si-Si結合,5配位Si等の欠陥構造が観察された。
水晶を接続した界面原子構造モデルに対して加熱・急冷計算を行い、アモルファスSiO/SiC界面原子構造を生成した。なお、加熱・急冷計算には第一原理分子動力学計算コードであるVASPを用いた。生成された界面構造モデルでは、従来モデルにおいて想定されていた、Si原子が界面にある2つないしは3つのSi-O結合をまとめる界面接続モデル以外にも、Si原子が一つのSi-O結合にのみ接続する構造ができていた。加えて従来モデルでは想定されていなかった、Siダングリングボンド,Si-Si結合,5配位Si等の欠陥構造が観察された。
no abstracts in English
使用言語 |
: | Japanese |
|---|---|---|
掲載資料名 |
: | |
巻 |
: | |
号 |
: | |
ページ数 |
: | |
発行年月 |
: | |
発表会議名 |
: | |
開催年月 |
: | 2009/03 |
開催都市 |
: | つくば |
開催国 |
: | 日本 |
キーワード |
: | no keyword |
論文URL |
: |
|
|---|---|---|
論文解説記事 |
: | |
受委託・共同研究相手機関 |
: |
Access |
: |
- Accesses |
|---|---|---|
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
 被引用回数:
被引用回数: