


 O)
O)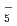 and (DO)
and (DO)Takayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Motegi, Haruki*; Shiga, Motoyuki 
![]()

Path-integral molecular dynamics simulations have been performed for water anion clusters,(HO) and (DO), on the basis of a semiempirical one-electron pseudopotential polarization model. Due to larger zero-point vibrational amplitudes for H atoms than that of D atoms, hydrogen-bond lengths in the (HO) cluster are slightly larger than those in (DO). The distribution of the vertical detachment energies for (HO) also show a broader feature than that for (DO). The present simulations thus demonstrate the importance of nuclear quantum effects in water anion clusters.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 482 |
Number |
: | 4-6 |
Pages |
: | p.195 - 200 |
Publication Year/Month |
: | 2009/12 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:28.06 Category:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
