


 O)
O)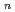 (
(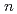 = 1-7) clusters on semi-empirical PM6 potential energy surfaces
= 1-7) clusters on semi-empirical PM6 potential energy surfacesYoshikawa, Takehiro*; Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Shiga, Motoyuki 
![]()

Path-integral molecular dynamics simulations for the hydrogen-bonded glycine-(HO) ( = 1-7) clusters have been carried out using an on-the-fly direct dynamics technique at the semiempiricalPM6 level of theory. In the case of smaller clusters with = 1-3, the simulations show that the clusterstructure takes exclusively the hydrogen-bonded complex between a canonical neutral glycine and a water cluster moiety. In contrast, it was found that proton-exchange processes effectively occur between the COOH carboxylic group and water cluster moiety for = 4-6 clusters although the overall structures are the complex between a neutral glycine and water clusters. In the case of the = 7 cluster, glycine preferentially takes a zwitterionic form having NH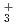 and COO
and COO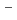 functional groups.
functional groups.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 365 |
Number |
: | 1-2 |
Pages |
: | p.60 - 68 |
Publication Year/Month |
: | 2009/12 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:46.32 Category:Chemistry, Physical |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
