


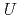 and
and 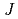 within a Pseudopotential formalism; Application to FeO and LaVO
within a Pseudopotential formalism; Application to FeO and LaVO
Hamada, Tomoyuki*; Ono, Takashisa*; Maekawa, Sadamichi
A first-principles density theory (DFT)+U method based on the pseudopotential (PP) method was developled to enable 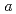 -
-  prediction of electronic structures of strongly-correlated electronic structures (SCES) systems. The method determines the radius of the localized electron orbitals of a SCES system and the Hubbard parameteres
prediction of electronic structures of strongly-correlated electronic structures (SCES) systems. The method determines the radius of the localized electron orbitals of a SCES system and the Hubbard parameteres 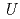 and
and 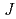 for the electrons and calculates the electronic structure of the system by using the rotationally-invariant DFT + U method and using the determined radius and parameters. The electronic structure of FeO in the anti-ferromagnetic state was calculated by using the method, and the method could describe the electronic structure of FeO correctly, producing an insulating ground state for FeO with an indirect band gap of 1.88 eV.
for the electrons and calculates the electronic structure of the system by using the rotationally-invariant DFT + U method and using the determined radius and parameters. The electronic structure of FeO in the anti-ferromagnetic state was calculated by using the method, and the method could describe the electronic structure of FeO correctly, producing an insulating ground state for FeO with an indirect band gap of 1.88 eV.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 62 |
Number |
: | 12 |
Pages |
: | p.2155 - 2159 |
Publication Year/Month |
: | 2013/06 |
Paper URL |
: |
|
Keywords |
: | no keyword |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:27.27 Category:Physics, Multidisciplinary |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
