


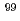 Ru M
Ru M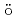 ssbauer parameters of ruthenium-nitrosyl complexes
ssbauer parameters of ruthenium-nitrosyl complexesKaneko, Masashi 
![]()
 ; Kato, Akane* ; Nakashima, Satoru*; Kitatsuji, Yoshihiro
; Kato, Akane* ; Nakashima, Satoru*; Kitatsuji, Yoshihiro
We applied density functional theory calculations to ruthenium-nitrosyl complexes, which are known to exist in high-level radioactive waste, to give a theoretical correlation between Ru Mssbauer spectroscopic parameters (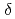 and
and 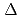
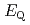 ) and ligand field strength (
) and ligand field strength ( ) for the first time. The structures of the series of complexes, [Ru(NO)L
) for the first time. The structures of the series of complexes, [Ru(NO)L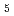 ] (L = Br
] (L = Br , Cl, NH
, Cl, NH , CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO
, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO )L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C
)L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C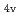 symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of
symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of 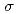 -donor and
-donor and 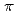 -acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
-acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
Language |
: | English |
|---|---|---|
Journal |
: | |
Volume |
: | 58 |
Number |
: | 20 |
Pages |
: | p.14024 - 14033 |
Publication Year/Month |
: | 2019/10 |
Paper URL |
: |
|
Keywords |
: |
Accesses |
: |
- Accesses |
|---|---|---|
InCites™ |
: |
Percentile:63.71 Category:Chemistry, Inorganic & Nuclear |
Altmetrics |
: |
[CLARIVATE ANALYTICS], [WEB OF SCIENCE], [HIGHLY CITED PAPER & CUP LOGO] and [HOT PAPER & FIRE LOGO] are trademarks of Clarivate Analytics, and/or its affiliated company or companies, and used herein by permission and/or license.
