


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
山本 風海; 金正 倫計; 林 直樹; Saha, P. K.; 田村 文彦; 山本 昌亘; 谷 教夫; 高柳 智弘; 神谷 潤一郎; 菖蒲田 義博; et al.
Journal of Nuclear Science and Technology, 59(9), p.1174 - 1205, 2022/09
被引用回数:6 パーセンタイル:84.97(Nuclear Science & Technology)J-PARC 3GeVシンクロトロン(RCS)は、最大1MWの大強度ビームを25Hzという早い繰り返しで中性子実験及び下流の主リングシンクロトロンに供給することを目的に設計された。2007年の加速器調整運転開始以降、RCSではビーム試験を通じて加速器の設計性能が満たされているかの確認を進め、必要に応じてより安定に運転するための改善を行ってきた。その結果として、近年RCSは1MWのビーム出力で連続運転を行うことが可能となり、共用運転に向けた最後の課題の抽出と対策の検討が進められている。本論文ではRCSの設計方針と実際の性能、および改善点について議論する。
近藤 恭弘; 浅野 博之*; 千代 悦司; 平野 耕一郎; 石山 達也; 伊藤 崇; 川根 祐輔; 菊澤 信宏; 明午 伸一郎; 三浦 昭彦; et al.
Proceedings of 28th International Linear Accelerator Conference (LINAC 2016) (Internet), p.298 - 300, 2017/05
J-PARC加速器の要素技術開発に必要な3MeV H リニアックを構築した。イオン源にはJ-PARCリニアックと同じものを用い、RFQは、J-PARCリニアックで2014年まで使用したものを再利用している。設置作業の後、2016年6月からRFQのコンディショニングを開始した。このRFQは様々な問題を克服し、なんとか安定運転に達していたが、2年間運転できなかったので再度コンディショニングが必要であった。現状定格のデューティーファクタでは運転できてはいないが、短パルスならばビーム運転可能となっている。この論文では、この3MeV加速器のコミッショニングと最初の応用例であるレーザー荷電変換試験の現状について述べる。
リニアックを構築した。イオン源にはJ-PARCリニアックと同じものを用い、RFQは、J-PARCリニアックで2014年まで使用したものを再利用している。設置作業の後、2016年6月からRFQのコンディショニングを開始した。このRFQは様々な問題を克服し、なんとか安定運転に達していたが、2年間運転できなかったので再度コンディショニングが必要であった。現状定格のデューティーファクタでは運転できてはいないが、短パルスならばビーム運転可能となっている。この論文では、この3MeV加速器のコミッショニングと最初の応用例であるレーザー荷電変換試験の現状について述べる。
澤邊 祐希*; 石山 達也; 高橋 大輔; 加藤 裕子; 鈴木 隆洋*; 平野 耕一郎; 武井 早憲; 明午 伸一郎; 菊澤 信宏; 林 直樹
Proceedings of 13th Annual Meeting of Particle Accelerator Society of Japan (インターネット), p.647 - 651, 2016/11
J-PARCでは実機の安定運転に必要なビームスクレーパ照射試験およびレーザ荷電変換試験を実施するために3MeVリニアックを再構築した。3MeVリニアックは、セシウム添加高周波駆動負水素イオン源(RFイオン源)から負水素イオンビームを取り出し、高周波四重極型リニアック(RFQ)で3MeVまでビームを加速する。3MeVリニアックを制御するには、加速器およびレーザから人への安全を確保する人的保護システム(PPS)、加速器構成機器を保護するための機器保護システム(MPS)、各機器の同期をとるタイミングステム、およびEPICSを用いた遠隔制御システムが重要となる。本発表では、これらの3MeVリニアック用制御システムについて報告する。
平野 耕一郎; 浅野 博之; 石山 達也; 伊藤 崇; 大越 清紀; 小栗 英知; 近藤 恭弘; 川根 祐輔; 菊澤 信宏; 佐藤 福克; et al.
Proceedings of 13th Annual Meeting of Particle Accelerator Society of Japan (インターネット), p.310 - 313, 2016/11
単位面積当たりの熱負荷を減らすため、67 のビーム入射角を有するビームスクレーパをJ-PARCリニアックのRFQとDTLの間のMEBTで使用している。67ビームスクレーパは粒子数1.47E22個のHビームによって照射された。レーザ顕微鏡を用いてスクレーパのビーム照射による損傷部を観察すると、高さ数百
のビーム入射角を有するビームスクレーパをJ-PARCリニアックのRFQとDTLの間のMEBTで使用している。67ビームスクレーパは粒子数1.47E22個のHビームによって照射された。レーザ顕微鏡を用いてスクレーパのビーム照射による損傷部を観察すると、高さ数百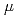 mの突起物が無数にあった。ビームスクレーパの耐電力を調べるため、3MeVリニアックを新たに構築した。2016年末にスクレーパ照射試験を実施する予定である。今回は、J-PARCリニアックのビームスクレーパの現状、及び、ビームスクレーパの照射試験に用いる3MeVリニアックについて報告する。
mの突起物が無数にあった。ビームスクレーパの耐電力を調べるため、3MeVリニアックを新たに構築した。2016年末にスクレーパ照射試験を実施する予定である。今回は、J-PARCリニアックのビームスクレーパの現状、及び、ビームスクレーパの照射試験に用いる3MeVリニアックについて報告する。
Yang, L.-W.*; 北尾 彰朗*; Huang, B.-C.*; 郷 信広*
Biophysical Journal, 107(6), p.1415 - 1425, 2014/09
被引用回数:18 パーセンタイル:54.3(Biophysics)In this study, a general linear response theory (LRT) is formulated to describe time-dependent and -independent protein conformational changes upon CO binding with myoglobin. Using the theory, we are able to monitor protein relaxation in two stages. The slower relaxation is found to occur from 4.4 to 81.2 picoseconds and the time constants characterized for a couple of aromatic residues agree with those observed by UV Resonance Raman (UVRR) spectrometry and time resolved X-ray crystallography. The faster "early responses", triggered as early as 400 femtoseconds, can be best described by the theory when impulse forces are used. The newly formulated theory describes the mechanical propagation following ligand-binding as a function of time, space and types of the perturbation forces. The "disseminators", defined as the residues that propagate signals throughout the molecule the fastest among all the residues in protein when perturbed, are found evolutionarily conserved and the mutations of which have been shown to largely change the CO rebinding kinetics in myoglobin.
甲斐 健師; 徳久 淳師*; 森林 健悟; 福田 祐仁; 河野 秀俊; 郷 信広*
Journal of the Physical Society of Japan, 83(9), p.094301_1 - 094301_5, 2014/09
被引用回数:1 パーセンタイル:11.67(Physics, Multidisciplinary)X線自由電子レーザー(XFEL)を利用した新たな単分子立体構造解析法が注目されており、この手法を実現するための最適条件の解明が期待されている。しかし、この手法では高強度のレーザーを使用するために、生じる分子の損傷が問題となる。本研究では単分子の損傷を考慮した上で、XFEL照射により得られる単分子の回折イメージング強度を様々な照射条件下において計算し、単分子立体構造解析を実現するために必要な照射条件について検討した。照射条件としてXFELの強度,エネルギー,パルス幅及び標的サイズをパラメータとし、標的を球状クラスターとした計算コードの開発を行った。本研究の成果により、XFEL照射による単分子の回折イメージング強度は入射X線のエネルギーが増加するとわずかに増加し、単分子の半径が400オングストローム以上になると照射強度に概ね比例することが分かった。XFEL照射条件を強度3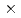 10
10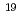 photons/mm
photons/mm 、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
徳久 淳師*; 新井 淳也*; 城地 保昌*; 大野 善之*; 亀山 豊久*; 山本 啓二*; 畑中 正行*; Gerofi, B.*; 島田 明男*; 黒川 原佳*; et al.
Journal of Synchrotron Radiation, 20(6), p.899 - 904, 2013/11
被引用回数:5 パーセンタイル:29.23(Instruments & Instrumentation)Single-particle coherent X-ray diffraction imaging using X-ray free electron laser has potential to reveal a three-dimensional structure of a biological supra-molecule at sub-nano meter resolution. In order to realize this method, it is necessary to analyze as many as one million noisy X-ray diffraction patterns, each for an unknown random target orientation. To cope with the severe quantum noise we need to classify patterns according to their similarities and average similar patterns to improve the S/N ratio. We developed a high-speed scalable scheme to carry out classification on the K computer, a 10PFLOPS supercomputer at RIKEN Advanced Institute for Computational Science. It is designed to work on the real time basis with the experimental diffraction pattern collection at the X-ray free electron laser facility SACLA so that the result of classification can be feed-backed to optimize experimental parameters during the experiment. We report the present status of our effort of developing the system and also a result of application to a set of simulated diffraction patterns. We succeeded in classification of about one million diffraction patterns by running 255 separate one-hour jobs on 385-node mode.
石山 達也; 伊藤 雄一; 菊澤 信宏; 鈴木 隆洋; 大井 元貴; 明午 伸一郎; 酒井 健二; 加藤 裕子
Proceedings of 9th Annual Meeting of Particle Accelerator Society of Japan (インターネット), p.714 - 716, 2013/08
J-PARCでは、MLFとMRの両施設へのビーム打ち分け時に、MLFにてMPSが発報すると全ビームが停止となり、正常運転状態にあるMRへのビーム供給も停止してしまう。特にミュオンターゲット移動時にはMPSが必ず発報する仕様となっているため、ビーム行先をMRのみに切り替えてターゲットを移動する運用を行っていた。このビーム行先切り替えの際にも全ビーム停止となる。これらによるMR運転稼働率の低下が問題となっていた。そこで、現状のMLFのMPS信号をMLFインヒビットとして扱い、MLFインヒビット発報時には、MLF行きのビームのみを停止し、MR運転稼働率の向上を図ることとした。勿論、MLFインヒビットを実現するにあたり、安全性を保つことが必須である。今回新たに、MLFインヒビット発報中にMLFにてビーム検出がされた際に、通常のMPSを発報し全ビーム停止を行うロジックを組み込むことで、稼働率向上、安全性確保を両立したMLFインヒビットシステムの構築を行った。
徳久 淳師*; 高 潤一郎*; 河野 秀俊; 郷 信広*
Acta Crystallographica Section A, 68(3), p.366 - 381, 2012/05
被引用回数:20 パーセンタイル:81.77(Chemistry, Multidisciplinary)A new two-step algorithm is developed for reconstructing three-dimensional diffraction intensity of a globular biological macromolecule from many experimentally measured quantum-noise limited two-dimensional (2D) X-ray laser diffraction patterns, each for unknown orientation. First step is a classification of 2D patterns into groups according to similarity of direction of incident X-ray with respect to the molecule and an averaging within each group to reduce the noise. Second is a detection of common intersecting circles between the signal-enhanced 2D patterns to identify their mutual location in the 3D wave-number space. The newly developed algorithm enables to detect signal for classification in such a noisy experimental photon-count data as low as  0.1 photons per effective pixel. Wavenumber of such a limiting pixel determines the attainable structural resolution. From this fact, resolution attainable by this new method of analysis as well as two important experimental parameters, the number of 2D patterns to be measured (load for detector) and the number of pairs of 2D patterns to be analyzed (load for computer), are derived as a function of intensity of incident X-ray and quantities characterizing the target molecule.
0.1 photons per effective pixel. Wavenumber of such a limiting pixel determines the attainable structural resolution. From this fact, resolution attainable by this new method of analysis as well as two important experimental parameters, the number of 2D patterns to be measured (load for detector) and the number of pairs of 2D patterns to be analyzed (load for computer), are derived as a function of intensity of incident X-ray and quantities characterizing the target molecule.
樋口 真理子; 藤井 淳平*; 米谷 佳晃; 北尾 彰朗*; 郷 信広*
Journal of Structural Biology, 173(1), p.20 - 28, 2011/01
被引用回数:2 パーセンタイル:6.15(Biochemistry & Molecular Biology)ヌクレオシドdGTPと損傷したヌクレオシド8oxodGTPは構造が似ているが、修復酵素MutTは結合力の大きな差によって両者を区別している。本研究では分子動力学シミュレーションの手法を用いて、基質の小さな差を増幅し大きな結合力の差として認識する分子的な仕組みを解明した。MutTはdGMPとの結合時は基質周辺のループが開いている状態が安定だが、8oxodGMPとの結合時は基質周辺のループが閉じている状態が安定であることがわかった。このような結合構造の違いが結合力の大きな差を生み出していると考えられる。さらに、基質のH7とMutTの119番目の残基Asnをつなぐ水素結合が結合構造の違いに重要な役割を果たしていることがわかった。
坂中 章悟*; 吾郷 智紀*; 榎本 収志*; 福田 茂樹*; 古川 和朗*; 古屋 貴章*; 芳賀 開一*; 原田 健太郎*; 平松 成範*; 本田 融*; et al.
Proceedings of 11th European Particle Accelerator Conference (EPAC '08) (CD-ROM), p.205 - 207, 2008/06
コヒーレントX線,フェムト秒X線の発生が可能な次世代放射光源としてエネルギー回収型リニアック(ERL)が提案されており、その実現に向けた要素技術の研究開発が日本国内の複数研究機関の協力のもと進められている。本稿では、ERL放射光源の研究開発の現状を報告する。
郷 信広
Progress of Theoretical Physics Supplement, (170), p.198 - 213, 2008/01
This is a record of my lecture given at the occasion of Yukawa-Tomonaga Centennial Symposium. At first I will mention very briefly how Yukawa contributed to the development of biophysics in Japan. Then I will be concerned with the relationship between physics and biology by discussing various aspects of protein. How far and in what sense can physics approach the essence of protein? In what aspects are something beyond physics important?
藤井 聡*; 河野 秀俊; 竹中 繁織*; 郷 信広; 皿井 明倫*
Nucleic Acids Research, 35(18), p.6063 - 6074, 2007/09
被引用回数:98 パーセンタイル:86.53(Biochemistry & Molecular Biology)タンパク質がDNAを認識するには、DNAとの水素結合や静電相互作用による直接的な認識とDNAの構造特性に由来した間接的な認識がある。われわれは、後者の間接的な認識を定量化するために、DNAの構造特性を分子動力学計算によって調べた。結果、これまで2塩基対で特徴づけられていた構造は、その特性を記述するのに不十分であること、塩基対の配列依存性が2つ先の塩基対までかなり影響することなどを示した。さらに、間接認識ポテンシャルを作成し、それが細胞内で見られるDNAのヌクレオソーム構造形領域を予測できることを示した。
Gong, X.*; 中村 建介; 由良 敬; 郷 信広
IEEE Transactions on Information Technology in Biomedicine, 11(4), p.428 - 434, 2007/07
被引用回数:3 パーセンタイル:27.84(Computer Science, Information Systems)グリッドコンピューティングの出現により、バイオインフォマティクスで普通に用いられる公共データベース,解析ツール及びワークフローを共有しながら研究をすることが可能となってきている。しかし、バイオインフォマティクス研究者にとっては、グリッドアプリケーションはまだまだ困難である。ここでは、バイオインフォマティクス解析をグリッド上で可能とするシステム(BAAQ)を紹介し、公共データベースと解析ツール及びワークフローの共有化の可能性を議論する。特にワークフローの簡単な構築方法と、今までに構築したワークフローの中からどのようにして必要なワークフローを検索するかを示す。そのうえで実問題への適用例を示す。
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2005 - March 2006, p.237 - 240, 2007/01
地球シミュレータは従来にはない大規模生体超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子の生体超分子系を扱う大規模な分子動力学シミュレーションシステムSCUBA(旧名PABIOS)を開発している。SCUBAはPPM計算法,系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,系の原子分布異方性により引き起こされる並列化効率の悪化を防ぐための動的ロードバランスなど、最新のアルゴリズムを採用した計算性能に優れたシミュレーションシステムである。本年度は、約55万原子の系(RuvAB-Holliday分岐DNA)について分子動力学シミュレーションを実行した結果、地球シミュレータで360個のプロセッサを用いてもSCUBAはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして、ホリデイジャンクションモデル(RuvA-Holliday分岐DNA)の長時間分子動力学シミュレーションを実行することで、Holliday分岐DNA結合タンパク質RuvAがDNAを組み換えるメカニズムを分子レベルで明らかにすることができた。
米谷 佳晃*; 河野 秀俊; 藤井 聡*; 皿井 明倫*; 郷 信広
Molecular Simulation, 33(1-2), p.103 - 107, 2007/01
被引用回数:5 パーセンタイル:16.62(Chemistry, Physical)5'AATT3'と5'TTAA3'の2種類の塩基配列のDNAについて分子動力学シミュレーションを行い、構造変化と水和の関係を調べた。シミュレーションから、5'AATT3'では、DNAは構造変化しにくく、水和水は構造化しやすいが、5'TTAA3'では、DNAは構造変化しやすく、水和水は構造化しやすいことが明らかになった。この結果に基づいてDNAの構造変化と水和の関係について議論した。
Kim, O. T. P.*; 由良 敬; 郷 信広
Nucleic Acids Research, 34(22), p.6450 - 6460, 2006/12
被引用回数:97 パーセンタイル:85.03(Biochemistry & Molecular Biology)タンパク質とRNAとの相互作用は、RNAの転写,加工,修復などの場面でみられる。RNAはタンパク質のどのような部分と相互作用するのかよく研究されていない。そこで、86個の原子分解能タンパク質-RNA相互作用構造データを用いて、相互作用部位の傾向を調べた。特にわれわれは新しい統計解析法としてタンパク質表面にあるアミノ酸残基のペアがどのようにRNAと相互作用するのかを解析した。これで得られた統計量を利用して、タンパク質の表面のどの部分にRNAが相互作用するかを推定することもかなり高い精度でできるようになった。
中川 洋; 片岡 幹雄*; 城地 保昌*; 北尾 彰朗*; 柴田 薫; 徳久 淳師*; 筑紫 格*; 郷 信広
Physica B; Condensed Matter, 385-386(2), p.871 - 873, 2006/11
被引用回数:13 パーセンタイル:52.05(Physics, Condensed Matter)スタフィロコッカルヌクレアーゼを用いてタンパク質のボソンピークの水和との関連を調べた。ボソンピークは合成高分子,ガラス性物質,アモルファス物質に共通に見られるものであるが、その起源は十分には理解されていない。ボソンピークに寄与する運動は調和振動である。水和によりピークの位置は高周波数側にシフトし、振動の力学定数は増加した。このことはタンパクのエネルギー地形が変化したことを示す。タンパク質が水和することでエネルギー地形がより凸凹になり、極低温ではエネルギー極小に振動がトラップされる。タンパク質のボソンピークの起源はこのエネルギー地形の凹凸と関係しているかもしれない。
北尾 彰朗*; 米倉 功治*; 米倉 さおり*; Samatey, F.*; 今田 勝巳*; 難波 啓一*; 郷 信広
Proceedings of the National Academy of Sciences of the United States of America, 103(13), p.4894 - 4899, 2006/03
被引用回数:45 パーセンタイル:63.3(Multidisciplinary Sciences)細菌べん毛繊維は一種類の蛋白質フラジェリンが会合してできた生体超分子であり、細菌の運動はべん毛モーターのトルクによって誘起されるべん毛繊維の左巻き-右巻き超らせん構造転移によって制御される。われわれは、大規模分子動力学シミュレーションによってさまざまな実験データを満足する原子レベルの超らせん構造を構築するとともに、構造多型の分子メカニズムを解明することに成功した。超らせん構造構築のメカニズムには以下の3種類の相互作用が鍵となることがわかった。パーマネント相互作用はさまざまな超らせん構造において常に保たれる。スライディング相互作用は可変な疎水性及び親水性アミノ酸残基ペアの間で形成され、大きなエネルギー変化なしに蛋白質サブユニット間のずれを許容する。スイッチ相互作用の形成と解消はサブユニット間相互作用とサブユニット内相互作用をそれぞれ安定化する。われわれは構造多型の原因は両者の相互作用のフラストレーションであると結論付けた。超らせん構造間の転移は「変形=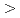 緩和」機構によって起こる。すなわち、繊維構造は幾何学的に急速に変形させられ、その後相互作用を再構成しながら徐々にエネルギー的準安定状態へ緩和することを明らかにした。
緩和」機構によって起こる。すなわち、繊維構造は幾何学的に急速に変形させられ、その後相互作用を再構成しながら徐々にエネルギー的準安定状態へ緩和することを明らかにした。
石田 恒; 樋口 真理子; 米谷 佳晃*; 叶野 琢磨; 城地 保昌*; 北尾 彰朗*; 郷 信広
Annual Report of the Earth Simulator Center April 2004 - March 2005, p.241 - 246, 2005/12
地球シミュレータは従来にはない大規模超分子系の分子動力学シミュレーションを可能とする計算能力を持つ。そこで、われわれは数百万原子のシステムを扱う大規模な分子シミュレーション(PABIOS)を開発している。PABIOSは空間分割法を用いており高い並列化効率を持つ。PABIOSは系のエネルギー,温度,圧力を一定に保つさまざまな時間積分アルゴリズム,原子結合長を固定することにより時間ステップの増大を可能とするSHAKE, RATTLEアルゴリズムなどの高精度アルゴリズムを採用した計算性能に優れたシミュレーションシステムである。今回、系の原子分布の異方性による並列化効率の悪化がおこらないように、動的ロードバランスを開発した。ホリデイ分岐DNAとDNA結合蛋白質RuvA4量体の複合体が水中に存在する系(全系16万6千原子)について分子動力学シミュレーションを実行した結果、地球シミュレータで120個のプロセッサを用いてもPABIOSはベクトル化率95%以上,並列化効率50%以上の優れた性能を達成することに成功した。そして計算速度も昨年度と比べて約2倍程度向上した。
