


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
中川 洋; 米谷 佳晃*; 中島 健次; 河村 聖子; 菊地 龍弥*; 稲村 泰弘; 片岡 幹雄*; 河野 秀俊*
JPS Conference Proceedings (Internet), 33, p.011101_1 - 011101_6, 2021/03
5'CGCG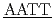 CGCG'3 and 5'CGCG
CGCG'3 and 5'CGCG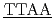 CGCG'3のDNAについて、軽水と重水のコントラストを利用した中性子準弾性散乱による水和水ダイナミクスを測定した。この2つのDNAは計算機によってそれぞれ硬い分子と柔らかい分子であることが分かっている。どちらの配列も約240KにDNAと水和水のどちらも動力学転移が観測された。転移温度以上では、水和水の平均自乗変位は硬い配列の方が小さかった。また水和水の緩和時間は硬いほうが長かった。ピコ秒時間スケールの水和水ダイナミクスは配列依存的なDNAの硬さと関係していることを示唆した。
CGCG'3のDNAについて、軽水と重水のコントラストを利用した中性子準弾性散乱による水和水ダイナミクスを測定した。この2つのDNAは計算機によってそれぞれ硬い分子と柔らかい分子であることが分かっている。どちらの配列も約240KにDNAと水和水のどちらも動力学転移が観測された。転移温度以上では、水和水の平均自乗変位は硬い配列の方が小さかった。また水和水の緩和時間は硬いほうが長かった。ピコ秒時間スケールの水和水ダイナミクスは配列依存的なDNAの硬さと関係していることを示唆した。
池部 仁善; 桜庭 俊*; 河野 秀俊
PLOS Computational Biology, 12(3), p.e1004788_1 - e1004788_13, 2016/03
被引用回数:45 パーセンタイル:90.91(Biochemical Research Methods)Acetylation of lysine residues in histone tails is associated with gene transcription. Because histone tails are structurally flexible and intrinsically disordered, it is difficult to experimentally determine the tail conformations and the impact of acetylation. In this work, we performed simulations to sample H3 tail conformations with and without acetylation. The results show that irrespective of the presence or absence of the acetylation, the H3 tail remains in contact with the DNA and assumes an alpha-helix structure in some regions. Acetylation slightly weakened the interaction between the tail and DNA and enhanced alpha-helix formation, resulting in a more compact tail conformation. We inferred that this compaction induces unwrapping and exposure of the linker DNA, enabling DNA-binding proteins (e.g., transcription factors) to bind to their target sequences. In addition, our simulation also showed that acetylated lysine was more often exposed to the solvent, which is consistent with the fact that acetylation functions as a post-translational modification recognition site marker.
河野 秀俊; 白山 一義*; 有村 泰宏*; 立和名 博昭*; 胡桃坂 仁志*
PLOS ONE (Internet), 10(3), p.e0120635_1 - e0120635_15, 2015/03
被引用回数:27 パーセンタイル:66.27(Multidisciplinary Sciences)The dynamics of nucleosomes containing either canonical H3 or its centromere-specific variant CENP-A were investigated using molecular dynamics simulations. The simulations showed that the histone cores were structurally stable during simulation periods of 100 ns and 50 ns, while DNA was highly flexible at the entry and exit regions and partially dissociated from the histone core. In particular, approximately 20-25 bp of DNA at the entry and exit regions of the CENP-A nucleosome exhibited larger fluctuations than DNA at the entry and exit regions of the H3 nucleosome. Our detailed analysis clarified that this difference in dynamics was attributable to a difference in two basic amino acids in the N helix; two arginine (Arg) residues in H3 were substituted by lysine (Lys) residues at the corresponding sites in CENP-A. The difference in the ability to form hydrogen bonds with DNA of these two residues regulated the flexibility of nucleosomal DNA at the entry and exit regions. Our exonuclease III assay consistently revealed that replacement of these two Arg residues in the H3 nucleosome by Lys enhanced endonuclease susceptibility, suggesting that the DNA ends of the CENP-A nucleosome are more flexible than those of the H3 nucleosome. This difference in the dynamics between the two types of nucleosomes may be important for forming higher order structures in different phases.
N helix; two arginine (Arg) residues in H3 were substituted by lysine (Lys) residues at the corresponding sites in CENP-A. The difference in the ability to form hydrogen bonds with DNA of these two residues regulated the flexibility of nucleosomal DNA at the entry and exit regions. Our exonuclease III assay consistently revealed that replacement of these two Arg residues in the H3 nucleosome by Lys enhanced endonuclease susceptibility, suggesting that the DNA ends of the CENP-A nucleosome are more flexible than those of the H3 nucleosome. This difference in the dynamics between the two types of nucleosomes may be important for forming higher order structures in different phases.
甲斐 健師; 徳久 淳師*; 森林 健悟; 福田 祐仁; 河野 秀俊; 郷 信広*
Journal of the Physical Society of Japan, 83(9), p.094301_1 - 094301_5, 2014/09
被引用回数:1 パーセンタイル:11.67(Physics, Multidisciplinary)X線自由電子レーザー(XFEL)を利用した新たな単分子立体構造解析法が注目されており、この手法を実現するための最適条件の解明が期待されている。しかし、この手法では高強度のレーザーを使用するために、生じる分子の損傷が問題となる。本研究では単分子の損傷を考慮した上で、XFEL照射により得られる単分子の回折イメージング強度を様々な照射条件下において計算し、単分子立体構造解析を実現するために必要な照射条件について検討した。照射条件としてXFELの強度,エネルギー,パルス幅及び標的サイズをパラメータとし、標的を球状クラスターとした計算コードの開発を行った。本研究の成果により、XFEL照射による単分子の回折イメージング強度は入射X線のエネルギーが増加するとわずかに増加し、単分子の半径が400オングストローム以上になると照射強度に概ね比例することが分かった。XFEL照射条件を強度3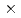 10
10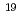 photons/mm
photons/mm 、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
中川 洋; 米谷 佳晃; 中島 健次; 河村 聖子; 菊地 龍弥; 稲村 泰弘; 片岡 幹雄; 河野 秀俊
Physical Review E, 90(2), p.022723_1 - 022723_11, 2014/08
被引用回数:10 パーセンタイル:55.34(Physics, Fluids & Plasmas)CGCGAATTCGCGとCGCGTTAACGCGの柔軟性の異なる二つの配列のDNAの分子シミュレーションと中性子準弾性散乱実験を行った。前者は硬く、後者は柔らかいことが知られている。両方の配列のDNAで、200-240Kに動力学転移が見られた。DNA配列依存的なダイナミクスを調べるために、転移温度以上でDNAと水和水のダイナミクスを分子シミュレーションと中性子準弾性散乱によって調べた。12merDNAの真ん中の4merについて、AATTはTTAAと比べて揺らぎの振幅が小さく、緩和時間が長いことが分かった。これはATステップの方が、TAステップよりも、速度論的に安定であることを示唆している。配列依存的な局所的な塩基対のダイナミクスは、DNAと水和水の間の水素結合ダイナミクスと相関がある。配列依存的なDNAの塩基対の揺らぎは動力学転移温度以上で現れる。これらの結果を総合すると、DNAの柔軟性は塩基対の局所的なダイナミクスと関係があり、DNAのマイナー溝に存在する水和水とカップルしていると結論付けた。
池部 仁善; 櫻庭 俊; 河野 秀俊
Journal of Computational Chemistry, 35(1), p.39 - 50, 2014/01
被引用回数:14 パーセンタイル:42.33(Chemistry, Multidisciplinary)A novel, efficient sampling method for biomolecules is proposed. The partial McMD was recently developed as a method that improved generalized ensemble (GE) methods to focus sampling only on a part of a system (GEPS); however, it was not tested well. We found that partial McMD did not work well for poly-lysine decapeptide and gave significantly worse sampling efficiency than a conventional GE. Herein, we elucidate the fundamental reason for this and propose a novel GEPS, adaptive lambda square dynamics (ALSD), which can resolve the problem faced when using partial McMD. We demonstrate that ALSD greatly increases the sampling efficiency over a conventional GE. We believe that ALSD is an effective method and is applicable to the conformational sampling of larger and more complicated biomolecule systems.
甲斐 健師; 徳久 淳師*; 河野 秀俊
Journal of the Physical Society of Japan, 82(11), p.114301_1 - 114301_5, 2013/11
被引用回数:1 パーセンタイル:11.42(Physics, Multidisciplinary)本研究では短パルス高強度X線レーザー照射による立体構造を考慮した単分子の損傷を時々刻々計算するために、分子動力学法に基づいた生体分子損傷のシミュレーションコードの開発を行った。本シミュレーションコードでは原子過程として光吸収電離過程,コンプトン散乱,オージェ過程のみならず、これまで分子動力学法をもとにした生体分子の損傷シミュレーションにおいて考慮されていなかった電場電離過程の効果を考慮した。本研究では標的としてリゾチームを選び、X線エネルギー12.4keV及びパルス幅5fsの条件で、パルス通過中の生体分子内の原子の平均束縛電子数及びイオンダイナミクスの計算を実行した。その結果、入射X線強度が110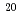 photons/mm以上になると、電場電離の効果によりリゾチーム分子の電離が急激に進行しはじめ、クーロン爆発は光電子が多数生成される110
photons/mm以上になると、電場電離の効果によりリゾチーム分子の電離が急激に進行しはじめ、クーロン爆発は光電子が多数生成される110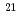 photons/mm程度の入射X線強度で誘発されることがわかった。
photons/mm程度の入射X線強度で誘発されることがわかった。
米谷 佳晃; 河野 秀俊
Journal of Physical Chemistry B, 117(25), p.7535 - 7545, 2013/06
被引用回数:15 パーセンタイル:37.47(Chemistry, Physical)DNA-binding proteins recognize DNA sequences with at least two different binding modes, specific and nonspecific. Experimental structures of such complexes provide us a static view of the bindings. However, it is difficult to reveal further mechanisms of their target-site search and recognition only from static information because the transition process between the bound and unbound states is not clarified by static information. What is the difference between specific and nonspecific bindings? Here we performed adaptive biasing force molecular dynamics simulations with the specific and nonspecific structures of DNA-Lac repressor complexes to investigate the dissociation process. The resultant free-energy profiles showed that the specific complex has a sharp, deep well consistent with tight binding, whereas the non-specific complex has a broad, shallow well consistent with loose binding. The difference in the well depth, 5 kcal/mol, was in fair agreement with the experimentally obtained value and was found to mainly come from the protein conformational difference, particularly in the C-terminal tail. Comparison of the free-energy barrier for sliding,  8.7 kcal/mol, and that for dissociation (at least 16 kcal/mol) calculated in this study suggests that sliding is much preferred to dissociation.
8.7 kcal/mol, and that for dissociation (at least 16 kcal/mol) calculated in this study suggests that sliding is much preferred to dissociation.
池田 思朗*; 河野 秀俊
統計数理, 61(1), p.135 - 146, 2013/06
現在、米国,欧州,日本及び韓国ではX線自由電子レーザーと呼ばれる装置が開発され、米国,日本では運用段階にある。現在、この装置によって初めて可能となったX線領域の波長を持つ高強度レーザー光を用いた、さまざまな実験が実施及び計画されている。本稿では、そうした実験のひとつ、X線回析を用いた生体単粒子の電子密度推定について説明し、2次元回析画像から2次元電子密度を推定する位相回復問題に対するわれわれの研究結果について解説する。
角南 智子; 河野 秀俊
PLOS ONE (Internet), 8(2), p.e56080_1 - e56080_12, 2013/02
被引用回数:12 パーセンタイル:30.66(Multidisciplinary Sciences)When a protein binds to DNA, a conformational change is often induced so that the protein will fit into the DNA structure. Therefore, quantitative analyses were conducted to understand the conformational changes in proteins. The results showed that conformational changes in DNA interfaces are more frequent than in non-interfaces, and DNA interfaces have more conformational variations in the DNA-free form. As expected, the former indicates that interaction with DNA has some influence on protein structure. The latter suggests that the intrinsic conformational flexibility of DNA interfaces is important for adjusting their conformation for DNA. The amino acid propensities of the conformationally changed regions in DNA interfaces indicate that hydrophilic residues are preferred over the amino acids that appear in the conformationally unchanged regions. This trend is true for disordered regions, suggesting again that intrinsic flexibility is of importance not only for DNA binding but also for interactions with other molecules. These results demonstrate that fragments destined to be DNA interfaces have an intrinsic flexibility and are composed of amino acids with the capability of binding to DNA. This information suggests that the prediction of DNA binding sites may be improved by the integration of amino acid preference for DNA and one for disordered regions.
河野 秀俊; 池田 思朗*
放射光, 26(1), p.38 - 43, 2013/01
SPring8に建設された短パルスかつ大強度のコヒーレントX線源は、生体高分子など単粒子での構造解析が期待されている。しかし、この従来の10億倍の輝度を持つ光を照射すると粒子は破壊されてしまうため、そうなる前の短時間(数fs以内)に回折パターンを観測しなくてはならない。そのため、測定される回折強度は弱いものになってしまう。われわれは、従来の方法よりも粗い回折パターンから位相回復できるベイズ統計にもとづいたアルゴリズムを開発したので、本稿で紹介する。
米谷 佳晃; 河野 秀俊
アンサンブル, 14(4), p.182 - 186, 2012/10
生体分子表面は複雑であり、それゆえ水のダイナミクスは多様である。水分子の生体分子表面滞在時間は、数psから数100psまでさまざまであるが、その違いはどのようにして生じるのであろうか。筆者らは、さまざまな塩基配列を持つDNAを対象にした分子動力学シミュレーションから、DNAと水の水素結合様式とDNA表面の構造揺らぎが、水分子の滞在時間に関係していることを明らかにした。そこでは、Laage-Hynesにより示された水素結合組換えのメカニズムとの接点も明らかになった。今後、タンパク質の場合なども含め、水分子の滞在時間を統一的に理解し、表現していくためには、これまで示唆されてきた表面の形状と電気的性質の影響も考慮しなければならない。
山崎 智*; 寺田 透*; 河野 秀俊; 清水 謙多郎*; 皿井 明倫*
Nucleic Acids Research, 40(17), p.e129_1 - e129_7, 2012/09
被引用回数:19 パーセンタイル:44.94(Biochemistry & Molecular Biology)Proteins recognize a specific DNA sequence not only through direct contact (direct readout) with base pairs but also through sequence-dependent conformation and/or flexibility of DNA (indirect readout). However, it is difficult to assess the contribution of indirect readout to the sequence specificity. What is needed is a straightforward method for quantifying its contributions to specificity. Using Bayesian statistics, we derived the probability of a particular sequence for a given DNA structure from the trajectories of molecular dynamics (MD) simulations of DNAs containing all possible tetramer sequences. Then, we quantified the specificity of indirect readout based on the information entropy associated with the probability. We tested this method with known structures of protein-DNA complexes. This method enabled us to correctly predict those regions where experiments suggested the involvement of indirect readout. The results also indicated new regions where the indirect readout mechanism makes major contributions to the recognition. The present method can be used to estimate the contribution of indirect readout without approximations to the distributions in the conformational ensembles of DNA, and would serve as a powerful tool to study the mechanism of protein-DNA recognition.
Adan, J.*; 吉井 周平*; 河野 秀俊; 宮田 真人*
Journal of Bacteriology, 194(12), p.3050 - 3057, 2012/06
被引用回数:9 パーセンタイル:24.08(Microbiology) is a parasitic bacterium that causes necrosis in the gills of freshwater fishes. This study examines the molecular structure of its surface variable protein, MvspI, composed of 2002 amino acids. MvspI was isolated from Mycoplasma cells by a biochemical procedure to 92% homogeneity. Gel filtration and analytical ultracentrifugation suggested that this protein is a cylinder-shaped monomer with axes of 66 and 2.7 nm. Rotary shadowing transmission electron microscopy of MvspI showed that the molecule is composed of two rods 30 and 45 nm long; the latter rod occasionally features a bulge. The electron microscopy with a monoclonal antibody and epitope mapping showed that the bulge end of molecular image corresponds to the C-terminus of amino acid sequence. Partial digestion by various proteases suggested that the N-terminal part, comprised of 697 amino acids, is flexible. Analysis of the predicted amino acid sequence showed that the molecule features an N-terminal signal sequence and 16 repeats of about 90 residues; 15 positions exist between residues 88 and 1479, and the other position is between residues 1725 and 1807. The amino acid sequence of MvspI was assigned onto a molecular image obtained by electron microscopy, based on the features from both analyses. The present study is the first to elucidate the molecular shape of a surface variable protein of Mycoplasma.
徳久 淳師*; 高 潤一郎*; 河野 秀俊; 郷 信広*
Acta Crystallographica Section A, 68(3), p.366 - 381, 2012/05
被引用回数:20 パーセンタイル:81.77(Chemistry, Multidisciplinary)A new two-step algorithm is developed for reconstructing three-dimensional diffraction intensity of a globular biological macromolecule from many experimentally measured quantum-noise limited two-dimensional (2D) X-ray laser diffraction patterns, each for unknown orientation. First step is a classification of 2D patterns into groups according to similarity of direction of incident X-ray with respect to the molecule and an averaging within each group to reduce the noise. Second is a detection of common intersecting circles between the signal-enhanced 2D patterns to identify their mutual location in the 3D wave-number space. The newly developed algorithm enables to detect signal for classification in such a noisy experimental photon-count data as low as 0.1 photons per effective pixel. Wavenumber of such a limiting pixel determines the attainable structural resolution. From this fact, resolution attainable by this new method of analysis as well as two important experimental parameters, the number of 2D patterns to be measured (load for detector) and the number of pairs of 2D patterns to be analyzed (load for computer), are derived as a function of intensity of incident X-ray and quantities characterizing the target molecule.
河野 秀俊; 今西 未来*; 根木 滋*; 辰谷 和弥*; 栄田 優衣*; 橋本 彩加*; 中山 千絵*; 二木 史朗*; 杉浦 幸雄*
FEBS Letters, 586(6), p.918 - 923, 2012/03
被引用回数:5 パーセンタイル:14.71(Biochemistry & Molecular Biology)We developed a rational scheme for designing DNA binding proteins. The scheme was applied for a zinc finger protein and the designed sequences were experimentally characterized with high DNA sequence specificity. Starting with the backbone of a known finger structure, we initially calculated amino acid sequences compatible with that expected structure and the designed fingers were experimentally confirmed their expected secondary structures. The DNA-binding function was then added to the designed finger by reconsidering a section of the amino acid sequence and computationally selecting amino acids to have the lowest protein-DNA interaction energy for the target DNA sequences. Among the designed proteins, one had a gap between the lowest and second lowest protein-DNA interaction energies that was sufficient to give DNA sequence-specificity.
池田 思朗*; 河野 秀俊
Optics Express (Internet), 20(4), p.3375 - 3387, 2012/02
被引用回数:10 パーセンタイル:46.91(Optics)In this paper, we propose the SPR (sparse phase retrieval) method, which is a new phase retrieval method for coherent X-ray diffraction imaging (CXDI). Conventional phase retrieval methods effectively solve the problem for high signal-to-noise ratio measurements, but would not be sufficient for single biomolecular imaging which is expected to be realized with femto-second X-ray free electron laser pulses. The SPR method is based on the Bayesian statistics. It does not need to set the object boundary constraint that is required by the commonly used hybrid input-output (HIO) method, instead a prior distribution is defined with an exponential distribution and used for the estimation. Simulation results demonstrate that the proposed method reconstructs the electron density under a noisy condition even some central pixels are masked.
米谷 佳晃; 河野 秀俊
Biophysical Chemistry, 160(1), p.54 - 61, 2012/01
被引用回数:20 パーセンタイル:54.2(Biochemistry & Molecular Biology)The lifetime during which a water molecule resides at the surface of a biomolecule varies according to the hydration site. What determines this variety of lifetimes? Despite many previous studies, there is still no uniform picture quantitatively explaining this phenomenon. Here we calculate the lifetime for a particular hydration pattern in the DNA minor groove, the water bridge, for various DNA sequences to show that the water-bridge lifetime varies from 1 to 300 ps in a sequence-dependent manner. We find that it follows 1/k (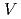
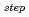 )
)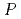
 , where and are two crucial factors, namely the probability of forming a specific hydrogen bond in which more than one donor atom participates, and the structural fluctuation of DNA, respectively. This relationship provides a picture of the water kinetics with atomistic detail and shows that water dissociation occurs when a particular hydrogen-bonding pattern appears.
, where and are two crucial factors, namely the probability of forming a specific hydrogen bond in which more than one donor atom participates, and the structural fluctuation of DNA, respectively. This relationship provides a picture of the water kinetics with atomistic detail and shows that water dissociation occurs when a particular hydrogen-bonding pattern appears.
米谷 佳晃; 河野 秀俊
揺らぎと生体機能, p.66 - 71, 2010/10
DNAの水和は、生命現象の根幹となる分子間相互作用の性質を理解するうえで重要である。近年、分子動力学シミュレーションにより、DNAのマイナーグルーブに現れる水和パターンに塩基配列依存性があること、また、水和水の挙動がDNAの構造揺らぎと密接に関係していることがわかってきた。本稿では、それらの知見について紹介するとともに、塩基配列によって異なる水和の性質がDNAとタンパク質,低分子の相互作用にどのように関連しているか議論する。
堀江 暁*; 富田 武郎*; 斉木 朝子*; 河野 秀俊; 高 ひかり*; 峯木 礼子*; 藤村 務*; 西山 千春*; 葛山 智久*; 西山 真*
Nature Chemical Biology, 5(9), p.673 - 679, 2009/09
被引用回数:43 パーセンタイル:67.5(Biochemistry & Molecular Biology)Although the latter part of the lysine biosynthesis, conversion of -aminoadipate (AAA) to lysine, of is similar to that of the arginine biosynthesis, enzymes homologous to ArgA and ArgJ are absent in the Thermus pathway. Since ArgA and ArgJ are known to modify the amino group of glutamate to avoid intramolecular cyclization of intermediates, their absence suggests an alternative N-modification system in the pathway. We reconstituted the conversion of AAA to lysine, and revealed that the amino group of AAA is modified with the 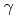 -carboxyl group of C-terminal Glu54 of a small protein, LysW, and the side-chain of AAA is converted to the lysyl side-chain in LysW-attached forms. Lysine is subsequently liberated from LysW/lysine-fusion. Recognition of the acidic globular domain of LysW by biosynthetic enzymes indicates that LysW acts as a carrier protein or protein scaffold of the biosynthetic enzymes in this system. This study reveals the previously unknown function of a small protein in the primary metabolism.
-carboxyl group of C-terminal Glu54 of a small protein, LysW, and the side-chain of AAA is converted to the lysyl side-chain in LysW-attached forms. Lysine is subsequently liberated from LysW/lysine-fusion. Recognition of the acidic globular domain of LysW by biosynthetic enzymes indicates that LysW acts as a carrier protein or protein scaffold of the biosynthetic enzymes in this system. This study reveals the previously unknown function of a small protein in the primary metabolism.
