


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Kuzubov, A. A.*; Kovaleva, E. A.*; Avramov, P. V.*; Kuklin, A. V.*; Mikhaleva, N. S.*; Tomilin, F. N.*; 境 誠司; 圓谷 志郎; 松本 吉弘*; 楢本 洋*
Journal of Applied Physics, 116(8), p.084309_1 - 084309_4, 2014/08
被引用回数:9 パーセンタイル:35.92(Physics, Applied)The interaction between carbon and BN nanotubes (NT) and transition metal Co and Ni supports was studied using electronic structure calculations. Several configurations of interfaces were considered, and the most stable ones were used for electronic structure analysis. All NT/Co interfaces were found to be more energetically favorable than NT/Ni, and conductive carbon nanotubes demonstrates lightly stronger bonding than semiconducting ones. The presence of contact-induced spin polarization was established for all nanocomposites. It was found that the contact-induced polarization of BNNT leads to the appearance of local conductivity in the vicinity of the interface while the rest of the nanotube lattice remains to be insulating.
 for lithium batteries proposed by
for lithium batteries proposed by  simulations
simulationsKuzubov, A. A.*; Fedorov, A. S.*; Eliseeva, N. S.*; Tomilin, F. N.*; Avramov, P.; Fedorov, D. G.*
Physical Review B, 85(19), p.195415_1 - 195415_4, 2012/05
被引用回数:43 パーセンタイル:81.93(Materials Science, Multidisciplinary)The absorption energy and diffusion rates of lithium atoms inside graphitelike boron carbide (BC) crystal are investigated by the pseudopotential density-functional method using generalized gradient approximation. It is shown that lithium may effectively intercalate this structure with the maximum lithium concentration corresponding to Li BC stoichiometry, which is threefold in comparison to lithium in graphite. The potential barrier values for lithium diffusion both at low and maximum concentration are about 0.19 eV, so lithium atoms inside the BC structure can move easily. These findings suggest that boron carbide looks like a good candidate as an anode material in lithium ion batteries.
BC stoichiometry, which is threefold in comparison to lithium in graphite. The potential barrier values for lithium diffusion both at low and maximum concentration are about 0.19 eV, so lithium atoms inside the BC structure can move easily. These findings suggest that boron carbide looks like a good candidate as an anode material in lithium ion batteries.
quantum nanodots and nanowiresAvramov, P.; Kuzubov, A. A.*; Fedorov, A. S.*; Sorokin, P. B.*; Tomilin, F. N.*; 前田 佳均
Physical Review B, 75(20), p.205427_1 - 205427_8, 2007/05
被引用回数:19 パーセンタイル:61.06(Materials Science, Multidisciplinary)本研究では、狭い界面を持つ5角形のSi/SiO量子ナノドット(QDs,直径1.6nm)及びナノワイヤ(NWs)を提案し、元となるシリコンの準安定構造(直径1.2nm)とともに、その原子構造と電子構造を、クラスター近似B3LYP/6-31G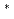 及び周期的境界条件(PBC)平面波近似(PW)擬ポテンシャル(PP)局所密度近似法を用いて計算した。最も小さい擬球状QD(Si
及び周期的境界条件(PBC)平面波近似(PW)擬ポテンシャル(PP)局所密度近似法を用いて計算した。最も小さい擬球状QD(Si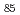 )の全状態密度(TDOS)はPBC PW PP LDAで計算した結晶性シリコンのTDOSによく一致した。伸長したSiQDsとSiNWsは金属的特性を持った電子構造を示す。しかし、表面に酸化層を持つとSi/SiOクラスターのTDOS中にバンドギャップができる。これらの粒子の価電子帯の上端と伝導帯の底は、核となるSiに由来した電子状態によって形成される。理論的に予測されるバンドギャップの幅は、Si/SiOクラスターの長さによって決定され、さらに、シリカに埋め込まれたナノシリコンのフォトルミネッセンススペクトルにおけるサイズ効果を高い精度で説明できる。
)の全状態密度(TDOS)はPBC PW PP LDAで計算した結晶性シリコンのTDOSによく一致した。伸長したSiQDsとSiNWsは金属的特性を持った電子構造を示す。しかし、表面に酸化層を持つとSi/SiOクラスターのTDOS中にバンドギャップができる。これらの粒子の価電子帯の上端と伝導帯の底は、核となるSiに由来した電子状態によって形成される。理論的に予測されるバンドギャップの幅は、Si/SiOクラスターの長さによって決定され、さらに、シリカに埋め込まれたナノシリコンのフォトルミネッセンススペクトルにおけるサイズ効果を高い精度で説明できる。
clusters, nanoparticles and nanowiresAvramov, P.; Kuzubov, A. A.*; Fedorov, A. A.*; Tomilin, F. N.*; Sorokin, P. B.*
no journal, ,
シリカのナノクラスターSi O
O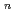 はSi/SiOナノ物質の前駆物質であり、その合成において重要な役割を果たす。本研究では、まず、SiO(m=2, 3, n=1-5)の生成と異性化について、その反応経路を6-31G(d)基底関数系を使った2次のMoller-Plesset摂動論(MP2)を用いて計算した。多くの異性化反応について遷移状態が存在することがわかったものの、分離された反応物からのクラスターの形成については、SiOとSiO
はSi/SiOナノ物質の前駆物質であり、その合成において重要な役割を果たす。本研究では、まず、SiO(m=2, 3, n=1-5)の生成と異性化について、その反応経路を6-31G(d)基底関数系を使った2次のMoller-Plesset摂動論(MP2)を用いて計算した。多くの異性化反応について遷移状態が存在することがわかったものの、分離された反応物からのクラスターの形成については、SiOとSiO についてのみ遷移状態が見いだされた。次に、密度汎関数法(B3LYP/6-31G)及び、周期的境界条件,擬ポテンシャル,平面波近似を用いた局所密度近似法を用いて、Si及びSi/SiOからなる量子ドット,ナノワイヤの電子構造を計算した。SiナノワイヤとSi量子ドットは、その多結晶性によりすべて金属的な電子状態を示すことがわかった。また、Siナノワイヤの表面に酸化によってSiOが存在するとSi/SiOナノワイヤの構造はすべて安定になり、SiOの存在によってバンドギャップ(
についてのみ遷移状態が見いだされた。次に、密度汎関数法(B3LYP/6-31G)及び、周期的境界条件,擬ポテンシャル,平面波近似を用いた局所密度近似法を用いて、Si及びSi/SiOからなる量子ドット,ナノワイヤの電子構造を計算した。SiナノワイヤとSi量子ドットは、その多結晶性によりすべて金属的な電子状態を示すことがわかった。また、Siナノワイヤの表面に酸化によってSiOが存在するとSi/SiOナノワイヤの構造はすべて安定になり、SiOの存在によってバンドギャップ( 1.4eV)を持つようになる。これらのSi/SiOナノ物質の価電子帯の上端と伝導帯の底は、おもにSiのp軌道で形成される。
1.4eV)を持つようになる。これらのSi/SiOナノ物質の価電子帯の上端と伝導帯の底は、おもにSiのp軌道で形成される。
