


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
甲斐 健師; 徳久 淳師*; 森林 健悟; 福田 祐仁; 河野 秀俊; 郷 信広*
Journal of the Physical Society of Japan, 83(9), p.094301_1 - 094301_5, 2014/09
被引用回数:1 パーセンタイル:11.67(Physics, Multidisciplinary)X線自由電子レーザー(XFEL)を利用した新たな単分子立体構造解析法が注目されており、この手法を実現するための最適条件の解明が期待されている。しかし、この手法では高強度のレーザーを使用するために、生じる分子の損傷が問題となる。本研究では単分子の損傷を考慮した上で、XFEL照射により得られる単分子の回折イメージング強度を様々な照射条件下において計算し、単分子立体構造解析を実現するために必要な照射条件について検討した。照射条件としてXFELの強度,エネルギー,パルス幅及び標的サイズをパラメータとし、標的を球状クラスターとした計算コードの開発を行った。本研究の成果により、XFEL照射による単分子の回折イメージング強度は入射X線のエネルギーが増加するとわずかに増加し、単分子の半径が400オングストローム以上になると照射強度に概ね比例することが分かった。XFEL照射条件を強度3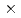 10
10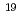 photons/mm
photons/mm 、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
、パルス幅1fs、エネルギー15.5keV、及び標的半径500オングストローム以上であると、単分子立体構造解析を行うための十分な回折イメージング強度が得られ、最適条件の存在が示された。
徳久 淳師*; 新井 淳也*; 城地 保昌*; 大野 善之*; 亀山 豊久*; 山本 啓二*; 畑中 正行*; Gerofi, B.*; 島田 明男*; 黒川 原佳*; et al.
Journal of Synchrotron Radiation, 20(6), p.899 - 904, 2013/11
被引用回数:5 パーセンタイル:29.23(Instruments & Instrumentation)Single-particle coherent X-ray diffraction imaging using X-ray free electron laser has potential to reveal a three-dimensional structure of a biological supra-molecule at sub-nano meter resolution. In order to realize this method, it is necessary to analyze as many as one million noisy X-ray diffraction patterns, each for an unknown random target orientation. To cope with the severe quantum noise we need to classify patterns according to their similarities and average similar patterns to improve the S/N ratio. We developed a high-speed scalable scheme to carry out classification on the K computer, a 10PFLOPS supercomputer at RIKEN Advanced Institute for Computational Science. It is designed to work on the real time basis with the experimental diffraction pattern collection at the X-ray free electron laser facility SACLA so that the result of classification can be feed-backed to optimize experimental parameters during the experiment. We report the present status of our effort of developing the system and also a result of application to a set of simulated diffraction patterns. We succeeded in classification of about one million diffraction patterns by running 255 separate one-hour jobs on 385-node mode.
甲斐 健師; 徳久 淳師*; 河野 秀俊
Journal of the Physical Society of Japan, 82(11), p.114301_1 - 114301_5, 2013/11
被引用回数:1 パーセンタイル:11.42(Physics, Multidisciplinary)本研究では短パルス高強度X線レーザー照射による立体構造を考慮した単分子の損傷を時々刻々計算するために、分子動力学法に基づいた生体分子損傷のシミュレーションコードの開発を行った。本シミュレーションコードでは原子過程として光吸収電離過程,コンプトン散乱,オージェ過程のみならず、これまで分子動力学法をもとにした生体分子の損傷シミュレーションにおいて考慮されていなかった電場電離過程の効果を考慮した。本研究では標的としてリゾチームを選び、X線エネルギー12.4keV及びパルス幅5fsの条件で、パルス通過中の生体分子内の原子の平均束縛電子数及びイオンダイナミクスの計算を実行した。その結果、入射X線強度が110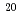 photons/mm以上になると、電場電離の効果によりリゾチーム分子の電離が急激に進行しはじめ、クーロン爆発は光電子が多数生成される110
photons/mm以上になると、電場電離の効果によりリゾチーム分子の電離が急激に進行しはじめ、クーロン爆発は光電子が多数生成される110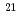 photons/mm程度の入射X線強度で誘発されることがわかった。
photons/mm程度の入射X線強度で誘発されることがわかった。
徳久 淳師*; 高 潤一郎*; 河野 秀俊; 郷 信広*
Acta Crystallographica Section A, 68(3), p.366 - 381, 2012/05
被引用回数:20 パーセンタイル:81.77(Chemistry, Multidisciplinary)A new two-step algorithm is developed for reconstructing three-dimensional diffraction intensity of a globular biological macromolecule from many experimentally measured quantum-noise limited two-dimensional (2D) X-ray laser diffraction patterns, each for unknown orientation. First step is a classification of 2D patterns into groups according to similarity of direction of incident X-ray with respect to the molecule and an averaging within each group to reduce the noise. Second is a detection of common intersecting circles between the signal-enhanced 2D patterns to identify their mutual location in the 3D wave-number space. The newly developed algorithm enables to detect signal for classification in such a noisy experimental photon-count data as low as  0.1 photons per effective pixel. Wavenumber of such a limiting pixel determines the attainable structural resolution. From this fact, resolution attainable by this new method of analysis as well as two important experimental parameters, the number of 2D patterns to be measured (load for detector) and the number of pairs of 2D patterns to be analyzed (load for computer), are derived as a function of intensity of incident X-ray and quantities characterizing the target molecule.
0.1 photons per effective pixel. Wavenumber of such a limiting pixel determines the attainable structural resolution. From this fact, resolution attainable by this new method of analysis as well as two important experimental parameters, the number of 2D patterns to be measured (load for detector) and the number of pairs of 2D patterns to be analyzed (load for computer), are derived as a function of intensity of incident X-ray and quantities characterizing the target molecule.
徳久 淳師; 城地 保昌*; 中川 洋; 北尾 彰朗*; 片岡 幹雄
Physical Review E, 75(4), p.041912_1 - 041912_8, 2007/05
被引用回数:20 パーセンタイル:67.12(Physics, Fluids & Plasmas)弾性非干渉性中性子散乱(EINS)では、散乱プロファイルの小角領域でガウス近似することで、揺らぎ幅の平均値である平均二乗変位(MSD)を見積もることがでる。一方、広角領域ではガウス近似からのずれ(非ガウス性)が観測される。非ガウス性には平均値以上の詳細な揺らぎ幅に関する情報が含まれており、非ガウス性に対しての解析法を確立することが望まれている。タンパク質ダイナミクスは非常に複雑で、非調和的であり、非等方的であり、かつ不均一である。そのため非ガウス性の起源に対して数種類の要因を考えることができる。非ガウス性の主要な起源を明らかにし、解析法を確立することを目的とした。核酸分解酵素であるStaphylococcal nucleaseの分子動力学計算から、EINSデータを再現した。非ガウス性を以下の3つの起源に分離し、非ガウス性を引き起こす種々の起源の寄与を見積もることに成功した。(1)揺らぎ幅の原子個々の不均一性の寄与(動的不均一性),(2)非等法性の寄与、及び(3)非調和性の寄与、に分離した。タンパク質全体では、さまざまなジャンプ距離を持つ原子が存在するため、非等方性及び非調和性の寄与は原子間で互いに打ち消し合い、EINSプロファイルへの寄与は結果的に小さくなることがわかった。つまり、非ガウス性の主要な起源は動的不均一性であり、タンパク質を試料としたEINS実験にあらわれる非ガウス性に対しては、動的不均一性の解析が適していることを明らかにした。
中川 洋; 徳久 淳師*; 上久保 裕生*; 城地 保昌*; 北尾 彰朗*; 片岡 幹雄*
Materials Science & Engineering A, 442(1-2), p.356 - 360, 2006/12
被引用回数:4 パーセンタイル:34.16(Nanoscience & Nanotechnology)中性子非干渉性弾性散乱と分子シミュレーションによって球状タンパク質の動的不均一性を調べた。中性子非干渉性弾性散乱のq依存性はガウス近似からのずれ、非ガウス性を示した。非調和性も非ガウス性に寄与するが、われわれは動的不均一性によって実際の散乱プロファイルの非ガウス性を説明することができた。分子シミュレーションにより、1meV程度の低いエネルギー分解能では非ガウス性はおもに動的不均一性に由来することを確認した。一方、非調和性の非ガウス性に対する寄与は10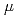 eV程度の高エネルギー分解能では無視できないが、それでも動的不均一性は非ガウス性の主な原因であった。
eV程度の高エネルギー分解能では無視できないが、それでも動的不均一性は非ガウス性の主な原因であった。
中川 洋; 片岡 幹雄*; 城地 保昌*; 北尾 彰朗*; 柴田 薫; 徳久 淳師*; 筑紫 格*; 郷 信広
Physica B; Condensed Matter, 385-386(2), p.871 - 873, 2006/11
被引用回数:13 パーセンタイル:52.05(Physics, Condensed Matter)スタフィロコッカルヌクレアーゼを用いてタンパク質のボソンピークの水和との関連を調べた。ボソンピークは合成高分子,ガラス性物質,アモルファス物質に共通に見られるものであるが、その起源は十分には理解されていない。ボソンピークに寄与する運動は調和振動である。水和によりピークの位置は高周波数側にシフトし、振動の力学定数は増加した。このことはタンパクのエネルギー地形が変化したことを示す。タンパク質が水和することでエネルギー地形がより凸凹になり、極低温ではエネルギー極小に振動がトラップされる。タンパク質のボソンピークの起源はこのエネルギー地形の凹凸と関係しているかもしれない。
中川 洋; 柴田 薫; 城地 保昌*; 徳久 淳師*; 片岡 幹雄*; 郷 信広
no journal, ,
タンパク質のダイナミクスは機能に重要である。タンパク質のダイナミクスの性質を理解するために、日本のつくばのKENSに設置されてあるLAM-40分光器を用いて非干渉性中性子非弾性散乱によりスタフィロコッカルヌクレアーゼのダイナミクスの水和と温度の影響を調べた。乾燥状態と重水水和及び軽水水和のタンパク質の非弾性散乱をさまざまな温度で測定した。乾燥状態の低温での非弾性散乱スペクトルには3meV付近にピークが観測された。このピークは水和することで高振動側へのシフトが観測された。このことは振動周波数分布が水和で変化することを示している。高温でのデバイワーラーファクターの異常な現象は、動力学転移と言われる平均自乗変位の増加を意味する。これは準弾性散乱の現象が現れることを示す。動力学転移温度よりも高温での運動の性質は準弾性散乱の解析により特徴付けられる。動力学転移は水和したタンパク質で顕著になる。このことは水和水がタンパク質のダイナミクスに強く影響していることを示している。水和水からの散乱プロファイルは軽水水和から重水水和のタンパク質の散乱を引くことによって得られる。水和水とタンパク質のダイナミクスの関係を議論する。
中川 洋; 城地 保昌*; 北尾 彰朗*; 柴田 薫; 徳久 淳師*; 郷 信広; 片岡 幹雄
no journal, ,
タンパク質のダイナミクスはその水和環境と強くカップルしている。タンパク質の動力学転移とボソンピークの水和との関係をスタフィロコッカルヌクレアーゼを用いて調べた。230K付近の動力学転移は水和タンパク質のみで観測される。タンパク質を動力学転移温度以下に下げると非調和的な運動が失われ、タンパク質の機能発現が抑制されることが示されている。動力学転移の水和量依存性を調べた。動力学転移は26%程度の高い水和量で観測された。過去の研究では約20%の水和量がタンパク質の機能に必要であるという報告がある。これは動力学転移がタンパク質の機能に重要であることを示している。一方、150K以下では低い水和量でもタンパク質の調和振動状態に影響を与えた。低温ではボソンピークが観測された。ボソンピークは合成高分子,ガラス,アモルファス物質などに共通して見られるが、その起源は十分には理解されていない。ピークに寄与する運動は調和振動である。水和するとピークの周波数は高振動にシフトし、低温での振動の力学定数は増加する。このことはタンパク質のエネルギー地形が変化したことを示す。タンパク質の水和はより凸凹したポテンシャル表面を作り出し、低温では振動運動はローカルミニマムにトラップされる。ボソンピークの起源はこの凸凹したエネルギー表面と関係があり、タンパク質の低振動モードの分布状態と関係している。
徳久 淳師; 石田 恒; 松本 淳; 河野 秀俊; 郷 信広
no journal, ,
これまでの構造解析は、ブラッグの散乱条件を用いた結晶構造解析が大部分を占め、構造を決定するには試料の結晶化が必要不可欠であった。しかし結晶化ができる生体高分子はごく一部である。このような背景のもと、X線自由電子レーザーを用いた、非結晶状態での生体高分子の立体構造決定法に大きな期待が寄せられている。本研究では計算科学的手法により、単分子X線構造解析の実現に向け、生体高分子の回転の影響及び、構造揺らぎの影響がスペックル散乱パターンに与える擾乱の程度を評価し、散乱パターンの分類法、及び立体構造構築法を確立することを目的とする。本年会では、単分子X線散乱データからの立体構造構築法の概要について報告する予定である。
徳久 淳師; 河野 秀俊
no journal, ,
The "zinc-finger" motif is an essential structure for proteins that bind DNA specifically. A transcription factor protein of Zif268, which consists of three zinc-finger motifs, recognizes the target sequence GCGTGGGCGT. How dose the Zif268 protein recognize the sequence? We present a quantitative analysis of the recognition mechanism of Zif268 using molecular dynamics simulations (MD). We performed MD simulations two different systems. One is the complex of Zif268 protein and target sequence DNA and another is the complex of Zif268 and non-target sequence (GCTATAAAAG) DNA. The result shows that the non-target sequence has larger structural entropy and smaller number of stable hydrogen bonds between DNA and protein than the target sequence. This difference leads to largely slide on the DNA surface of the finger 2 of Zif268 that binds non-target sequence by a foothold of ARG 40 and ARG44 residues.
徳久 淳師; 河野 秀俊
no journal, ,
マウスの発生初期に発現するタンパク質Zif268は、ジンクフィンガーモチーフ3つから構成されており、Gリッチな塩基配列を認識し結合する。本研究ではZif268の認識機構の解明を目的に、認識塩基配列と非認識塩基配列、2つの異なる系で分子動力学計算を行い、その結果を比較した。結果、タンパク質-DNA間の相対位置の変化が、非認識配列の系では認識配列の系に比べ大きいことがわかった。特に非認識配列の系においては、2つ目のフィンガー部位(F2)がDNA表面をすべるような運動を観測した。またDNAの構造エントロピーは非認識配列のほうが大きく、タンパク質-DNA間に形成される安定な水素結合が認識配列に比べ少ないことがわかった。以上のことから、両者間の運動性の違いは、タンパク質-DNA間に形成される水素結合に起因しており、このエンタルピックな効果によりZif268は特異的な認識を安定化していると考えられる。認識配列の系で特に安定な水素結合を形成した、Arg44, Asp46, His47の3つの残基が、F2のDNA認識において塩基配列特異性を担っていると考えられる。
徳久 淳師; 高 潤一郎; 森林 健悟; 乙部 智仁; 甲斐 健師; 中村 龍史; 福田 祐仁; 河野 秀俊; 郷 信広
no journal, ,
A promising tool for 3D-structural determination from a single-molecule without crystallized is X-ray free electron lasers (XFELs), which are now under construction in Europe, USA and Japan, independently. Our final goal is to develop a computational method for the determination of single-molecule 3D-strucures and to suggest feasible parameters of the target molecule and the XFEL device. When a molecule is irradiated by a coherent X-ray pulse with an ultrashort duration time and an extremely high intensity, a continuous diffraction pattern (2D speckle pattern) is observed. It is necessary to collect many 2D speckle patterns from different orientations of the molecule to obtain information sufficient for the structure determination. In the meeting, we present our strategy for determining 3D structures of biomolecules and suggest some critical parameters of the XFEL device and the target molecules required for realizing the 3D structure determination.
徳久 淳師; 高 潤一郎; 河野 秀俊; 郷 信広*
no journal, ,
New light sources of X-ray free-electron laser (XFEL) are under construction in Europe, USA and Japan. XFEL offers a new possibility in imaging single biological macromolecules. Our goal is to develop a computational method for the determination of molecule 3D structures from diffraction intensity data by XFEL. A straight forward procedure would be (1) to classify and average 2D diffraction patterns to improve the S/N ratio, (2) to obtain a 3D diffraction pattern by determining the relative positions of averaged-2D-images and (3) to retrieve the phases and construct the 3D structure using the over-sampling method. One of the big problems is the quantum noise resulting from the extremely weak intensity of elastically diffracted X-ray. Algorithms for structure determination must be developed to process the experimental data immersed in the quantum noise. In this meeting we report detailed procedures of analysis of experimental diffraction intensity data to be obtained 3D structure.
徳久 淳師*; 甲斐 健師; 高 潤一郎*; 河野 秀俊; 郷 信広*
no journal, ,
XFEL can potentially offer a new mean for single biomolecule 3D-imaging. Measured intensity, which is a 2D diffraction pattern per one observation without phase information is extremely weak even using XFEL because sample is a single molecule in a single shot and incident intensity of XFEL cannot make stronger than a certain intensity due to suppress molecule decay. Under such experimental conditions, we consequently have to use extremely weak signals effectively to reconstruct a 3D molecule structure using multiply measured many 2D diffraction patterns. We have developed a two-step algorithm for reconstructing a 3D diffraction intensity data from many experimentally measured, quantum-noise limited 2D patterns of single molecule with unknown orientation. Once a 3D diffraction intensity data of a molecule obtained, the 3D structure can be obtained by applying oversampling method to the data. The developed algorithm enables us to detect as low as 0.1 photons per an effective pixel as signal for the classification in such a noisy experimental photon-count data. Theoretically, wave-number of a pixel giving such a limiting photon-count defines the attainable structural resolution.
河野 秀俊; 池田 思朗*; 徳久 淳師*
no journal, ,
A new phase retrieval method for noisy X-ray diffraction imaging is proposed. Conventional phase retrieval methods effectively solve the problem for high signal-to-noise ratio measurements, but would not be sufficient for single biomolecular imaging which is expected to be realized with femto-second X-ray free electron laser pulses which is soon available at SPring8. The new phase retrieval method is based on the Bayesian statistical approach. The features of the method are that (1) it does not need to set the object boundary constraint that is required by the commonly used hybrid input-output (HIO) method, instead a sparse prior is used for the object estimation, and we call it SPR (sparse phase retrieval) method and (2) it is about 10 times robust to noise compared with HIO method. The simulation results demonstrate that the SPR method can reconstruct the object efficiently under a realistic condition.
Putra, E. G. R.*; 河野 秀俊; 徳久 淳師*; Bahrum, E. S.*; Patriati, A.*
no journal, ,
Molecular dynamics (MD) simulation of hen egg white lysozyme protein (4LZT, with and without disulphide bonds) and Ca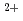 -binding protein calmodulin (1PRW, bent conformation and 1EXR, extended conformation) in solution have been explored using computer simulation. The dynamics of the proteins in aqueous phase at room temperature was produced for 5 nanosecond (ns) time-scale calculation. The trajectories generated from MD simulation were computed to produce the pair distribution function (PDF), the distribution of the neutron scattering length density as a function of atomic distances of a single molecule protein. Conclusively an inverse Fourier transform was applied to transform a pair-distribution function data into a theoretical small-angle neutron scattering (SANS) intensity against scattering vector (q) which can be experimentally evaluated. Several information such as the radius of gyration (Rg) and the shape of protein in solution can be obtained at every single time of simulation. In this study, the time-dependent structural changes in the extended conformation of Ca-binding protein calmodulin every 100 picosecond (ps) presented in through the pair-distribution function and SANS scattering profiles.
-binding protein calmodulin (1PRW, bent conformation and 1EXR, extended conformation) in solution have been explored using computer simulation. The dynamics of the proteins in aqueous phase at room temperature was produced for 5 nanosecond (ns) time-scale calculation. The trajectories generated from MD simulation were computed to produce the pair distribution function (PDF), the distribution of the neutron scattering length density as a function of atomic distances of a single molecule protein. Conclusively an inverse Fourier transform was applied to transform a pair-distribution function data into a theoretical small-angle neutron scattering (SANS) intensity against scattering vector (q) which can be experimentally evaluated. Several information such as the radius of gyration (Rg) and the shape of protein in solution can be obtained at every single time of simulation. In this study, the time-dependent structural changes in the extended conformation of Ca-binding protein calmodulin every 100 picosecond (ps) presented in through the pair-distribution function and SANS scattering profiles.
徳久 淳師*; 城地 保昌*; 河野 秀俊; 郷 信広*
no journal, ,
We proposed a method for classifying and assembling two-dimensional diffraction patterns to reconstruct the three-dimensional diffraction intensity functions. In the classification process, similarity of each pair of patterns is judged based on the correlation of corresponding pixels along the concentric circles When two diffraction patterns are similar to each other, a straight line with high correlations appears from the origin to a certain high scattering-angle region. Theoretically, the integrated value of the correlation value along this line is proportional to the extent of the similarity and can be used as threshold for the classification. This correlation line disappears with increasing scattering-angle due to the quantum noise and the structural irregularity. In our estimation, diffraction images of 106orders will be required to construct the 3D structure with 3 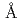 ; resolution for a molecule with a radius of 200 ; In this case, the necessary number of pattern comparisons is 10
; resolution for a molecule with a radius of 200 ; In this case, the necessary number of pattern comparisons is 10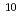 order. To achieve such a huge number of comparisons, we have to develop an automatic system. In this meeting, we will report how to deal with such a huge amount of data and our current status of the system.
order. To achieve such a huge number of comparisons, we have to develop an automatic system. In this meeting, we will report how to deal with such a huge amount of data and our current status of the system.
