


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 path integral Monte Carlo simulations for water trimer with electron correlation effects
path integral Monte Carlo simulations for water trimer with electron correlation effects藤田 貴敏*; 田中 成典*; 藤原 崇幸*; 草 将晃*; 望月 祐志*; 志賀 基之
Computational and Theoretical Chemistry, 997, p.7 - 13, 2012/10
水の三量体とは、凝集相にある水分子間の水素結合状態を特徴づけるモデルとして、実験と理論の両面から活発に研究が行われている系である。本論文では、この水三量体を対象として、電子相関効果を三次のM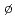 ller-Plesset摂動法まで考慮した高精度の第一原理経路積分モンテカルロ法を用いて、電子と原子核からなる系をすべて量子的に扱った第一原理シミュレーションを行った。具体的には、水三量体におけるO-O距離, O-H-O角度, H-OOO二面角など、水素結合を特徴づける構造因子を対象として、核の量子効果,熱的ゆらぎ効果,電子相関効果が与える影響について、系統だった解析を行い、これにより、計算で得られた水素結合構造の誤差を定量的に見積ることが可能となり、実験データとの直接比較ができるようになった。これらの成果は、水分子間の水素結合の基礎的理解を進めるものだが、高精度な原子分子レベルの計算手法の開発とその検証に該当し、原子力分野での応用が期待される。
ller-Plesset摂動法まで考慮した高精度の第一原理経路積分モンテカルロ法を用いて、電子と原子核からなる系をすべて量子的に扱った第一原理シミュレーションを行った。具体的には、水三量体におけるO-O距離, O-H-O角度, H-OOO二面角など、水素結合を特徴づける構造因子を対象として、核の量子効果,熱的ゆらぎ効果,電子相関効果が与える影響について、系統だった解析を行い、これにより、計算で得られた水素結合構造の誤差を定量的に見積ることが可能となり、実験データとの直接比較ができるようになった。これらの成果は、水分子間の水素結合の基礎的理解を進めるものだが、高精度な原子分子レベルの計算手法の開発とその検証に該当し、原子力分野での応用が期待される。
path integral Monte Carlo simulations for water trimer with electron correlation effects藤田 貴敏*; 田中 成典*; 藤原 崇幸*; 草 将晃*; 望月 祐志*; 志賀 基之
Computational and Theoretical Chemistry, 997, p.7 - 13, 2012/10
被引用回数:9 パーセンタイル:21.13(Chemistry, Physical)固体や液体等において、低原子量の元素から構成される化合物のダイナミクスを解析する場合には、原子核の量子ダイナミクスを考慮する必要が指摘されており、原子力分野をはじめとしてさまざまな分野において、それを考慮した新規のシミュレーション手法の開発等が議論されてきている。本発表では、この観点に立ち、電子と原子核からなる系をまるごと量子力学的に扱った第一原理経路積分モンテカルロシミュレーションにより、三量体の水クラスターの構造を精密に調べた結果を報告する。なお、採用した手法は、三次の多体摂動論に基づいた電子状態理論で求められた断熱ポテンシャル上のもとで、量子及び熱的効果を含めた分子構造の動的なゆらぎを求めることのできる極めて精度の高いシミュレーション手法であり、計算の結果、水クラスターの水素結合の柔軟性は、従来の第一原理計算から予測されていた結果よりも大きく、酸素間距離が大きな振幅で振動する様子が明らかになった。
岡本 穏治*; 望月 祐志*; 津島 悟*
Chemical Physics Letters, 373(1-2), p.213 - 217, 2003/05
被引用回数:8 パーセンタイル:24.96(Chemistry, Physical)4価トリウムイオンは、pHが3よりも大きい条件では容易に加水分解反応(Th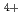 +4H
+4H O
O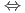 Th(OH)
Th(OH)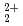 +2H
+2H O
O )を起こすことが古くから知られているが、反応の活性障壁,反応熱,電子分布の変化などの詳細については未知のまま残されていた。本研究では、非経験的分子軌道計算により、トリウムイオンと水の集合体から成るクラスターモデルを系統的に拡大させながら、反応をシミュレートした。計算から、反応が大きな発熱を伴うこと,始原系に近い遷移状態構造を持つこと,協調的に電子移動が起きることなどが示された。
)を起こすことが古くから知られているが、反応の活性障壁,反応熱,電子分布の変化などの詳細については未知のまま残されていた。本研究では、非経験的分子軌道計算により、トリウムイオンと水の集合体から成るクラスターモデルを系統的に拡大させながら、反応をシミュレートした。計算から、反応が大きな発熱を伴うこと,始原系に近い遷移状態構造を持つこと,協調的に電子移動が起きることなどが示された。
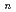 (
(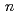 =1-4) by all-electron Dirac-Hartree-Fock calculations
=1-4) by all-electron Dirac-Hartree-Fock calculations望月 祐志*; 舘脇 洋*
Journal of Chemical Physics, 118(20), p.9201 - 9207, 2003/05
被引用回数:6 パーセンタイル:18.14(Chemistry, Physical)弗化物はアクチニドのポピュラーな化合物であるが、原子力エネルギー分野の主役であるUとPuの弗化物を例外として他の元素についてはほとんど知見が得られていない。しかし、ランタニドと比較すると後期のアクチニドでも+4価と取れるなど化学的な差異は知られており、計算によってこうした側面に切り込むことが出来れば、原子力工学・基礎無機化学にとって大きな意味が期待される。本研究では、CmとGdを例に取って、4弗化物までをDirac-Hartree-Fock計算によって系統的に調べた。本結果から、「CmF が合成可能なのに、GdFが不可能な理由」,中心金属の実効電子配置などの知見が世界で初めて明らかにされた。大規模並列計算としても、当該分野で過去最大のものである。
が合成可能なのに、GdFが不可能な理由」,中心金属の実効電子配置などの知見が世界で初めて明らかにされた。大規模並列計算としても、当該分野で過去最大のものである。
and Ac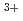 hydrate models
hydrate models望月 祐志*; 津島 悟*
Chemical Physics Letters, 372(1-2), p.114 - 120, 2003/04
被引用回数:13 パーセンタイル:39.19(Chemistry, Physical)アクチニドの代表的な4価イオンであるトリウム(Th)の水和モデルをDirac-Hartree-Fock法によって計算し、電子構造を詳細に調べた。比較のため同じ5f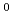 イオンであるAcのモデルも同様に計算した。Thの場合、空の6dや5f殻への水側からの電子供与が大きいが、合わせて準内殻の6pに「孔」が開き、これにより水側の電子構造も影響を受けていることがわかった。また、有効ポテンシャル近似に誘電体水和場法を組み合わせた計算も併せて行い、妥当なTh-O距離を得るにはバルクの水和場の考慮が重要であることも示した。
イオンであるAcのモデルも同様に計算した。Thの場合、空の6dや5f殻への水側からの電子供与が大きいが、合わせて準内殻の6pに「孔」が開き、これにより水側の電子構造も影響を受けていることがわかった。また、有効ポテンシャル近似に誘電体水和場法を組み合わせた計算も併せて行い、妥当なTh-O距離を得るにはバルクの水和場の考慮が重要であることも示した。
舘脇 洋*; 望月 祐志*
Theoretical Chemistry Accounts, 109(1), p.40 - 42, 2003/01
被引用回数:23 パーセンタイル:52.43(Chemistry, Physical)Dirac-Hartree-Fockを始め、4成分の相対論的分子軌道計算を行うにあたって、変分崩壊を起こしにくく、またいわゆる「基底関数重ね合わせ誤差」の少ない基底関数の選択は重要である。しかし、4成分計算は計算コストが高いために、非対称論系の基底に比して選択すべき候補も少なく、また検証も未だ進んでいない。本論文ではこうした状況をふまえ、キュリウムとその弗化物を例に取り、基底関数選択にあたっての注意点を考察し、簡潔にまとめた。
, f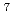 , and f
, and f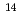 ions
ions望月 祐志*; 岡本 穏治*
Chemical Physics Letters, 359(3-4), p.331 - 336, 2002/06
被引用回数:2 パーセンタイル:5.55(Chemistry, Physical)3価のアクチノイドイオン(Ac,Cm,Lr)とランタノイドイオン(La,Gd,Lu)とアンモニア分子との錯体(アンミン錯体)について、Dirac-Hartree-Fock,2次摂動の4成分相対論計算を行った。金属-窒素間距離,安定化エネルギー,Mulliken密度などを評価し、それらを以前扱ったアクア(水)錯体と系統的に比較した。結果としてアンモニアの方がアクチノイドイオンとの「相性」が良いことがわかった。
O)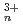 (n=1, 2, 4, 6) by all-electron Dirak-Hartree-Fock calculations
(n=1, 2, 4, 6) by all-electron Dirak-Hartree-Fock calculations望月 祐志*; 舘脇 洋*
Journal of Chemical Physics, 116(20), p.8838 - 8842, 2002/05
被引用回数:16 パーセンタイル:45.25(Chemistry, Physical)全電子のDirac-Hartree-Fock法を並列処理を駆使することによって、3価キュリウムイオンの6水和錯体モデルにまで適用した。さらに、蛍光スペクトルの評価のために閉殻完全CI計算も行った。同様の計算を等電子系のガドリニウムイオンについても用い、結果をキュリウムの場合と比較した。一連の計算により、水和の本質が配位結合による安定化であること、水側からイオンへ相当量の電子供与があること、結果として蛍光スペクトルが赤方レフトすること等が明らかになった。
望月 祐志*; 松村 昌幸*; 与倉 徹一*; 平原 幸男*; 今村 俊幸
Journal of Nuclear Science and Technology, 39(2), p.195 - 199, 2002/02
被引用回数:5 パーセンタイル:34.58(Nuclear Science & Technology)DIRACは、2電子積分から直接Fock行列を計算することにより大規模なDirac-Hartree-Fock計算が可能なソフトウェアである。積分の寄与を計算する際、コストの大きい交換項ではアドレスの衝突が起こるために単純なベクトル化はできない。今回、作業配列を導入することによりベクトル化を達成した。また、前段階のスクリーン処理も合わせてベクトル化した。ベクトル化率は、この結果90%を超え、Dirac-Hartree-Fockで2倍,線形応答では約3倍の加速が得られた。なお、この研究は地球シミュレータ用のソフトウェア整備の一環として行ったものである。
望月 祐志*; 舘脇 洋*
Chemical Physics, 273(2-3), p.135 - 148, 2001/11
被引用回数:23 パーセンタイル:58.82(Chemistry, Physical)4成分の相対論的分子軌道計算(Dirac-Hartree-Fock,Relativistic M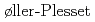 摂動)により、3価のランタノイド,アクチノイドイオンと水分子との錯体を系統的に調べ、イオン-水分子間距離,安定化エネルギーMulliken密度などを評価した。本計算から、相対論効果の大きさ、並びに相対論と電子相関の分離性が示されたほか、錯体における電子的相互作用の描像が明らかになった。
摂動)により、3価のランタノイド,アクチノイドイオンと水分子との錯体を系統的に調べ、イオン-水分子間距離,安定化エネルギーMulliken密度などを評価した。本計算から、相対論効果の大きさ、並びに相対論と電子相関の分離性が示されたほか、錯体における電子的相互作用の描像が明らかになった。
望月 祐志;  gren, H.*
gren, H.*
Chemical Physics Letters, 336(5-6), p.451 - 456, 2001/03
被引用回数:14 パーセンタイル:41.27(Chemistry, Physical)シリコン結晶から切り出し、水素終端化したクラスターモデルに対して、並列処理を援用する大規模な線形応答計算を行い、分極率並びに励起エネルギーを評価した。最大のSi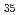 H
H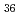 では静的分極率として3.87Å
では静的分極率として3.87Å が得られ、結晶シリコンの実験値3.64-3.76Åによく対応する。
が得られ、結晶シリコンの実験値3.64-3.76Åによく対応する。
望月 祐志*; 小出 洋; 今村 俊幸; 武宮 博*
Journal of Synchrotron Radiation, 8(Part.2), p.1003 - 1005, 2001/03
アデニンとグアニン、この2つのプリン塩基分子は、DNAを構成する重要な化合物だが、これまでその物性は原子価電子によるものがおもに取り上げられてきた。原子価電子は非局在性が高いため、原子の局所的情報を得るには不向きである。一方、内殻電子にかかわるX線吸収スペクトルは、Xの局在性故に各原子の局所的化学環境が調べられている。この研究では、プリン塩基の窒素K殻スペクトルを、プリン環への化学修飾、水和などを組み合わせた一連の系へのHF-STEX,RASSCF計算によりシミュレーションし、窒素原子K殻吸収端エネルギーがいかにシフトするか系統的に評価する。この計算の遂行には大規模な並列処理にするが、将来さらに大型の計算も可能とするよう、異機種計算機上での分散並列への対応も合わせて進めている。
武宮 博*; 今村 俊幸; 小出 洋; 樋口 健二; 辻田 祐一; 山岸 信寛*; 松田 勝之*; 上野 浩一*; 長谷川 幸弘*; 木村 俊哉; et al.
Proceedings of 4th International Conference on Supercomputing in Nuclear Applications (SNA 2000) (CD-ROM), 16 Pages, 2000/09
並列分散型の科学計算の開発及び実行環境を支援するために、STA(Seamless Thinking Aid)と呼ばれる計算環境を開発した。STAは、(1)各プログラム・コンポーネントの開発環境、(2)各々のコンポーネントをまとめて一つのアプリケーションに形成する機能、そして(3)アプリケーションを分散した計算資源に配分する機能等のツール群を提供する。STAの有用性を立証するために、われわれはいくつかの並列科学計算のアプリケーションを開発してきた。ここでは、これらのアプリケーションの特徴とSTAにおける構築法について述べる。
望月 祐志
JAERI-Data/Code 99-015, 21 Pages, 1999/03
 JAERI-Data-Code-99-015.pdf:0.84MB
JAERI-Data-Code-99-015.pdf:0.84MB
代替フロンは、塩素を含まないためフロンに比して「問題が少ない」とされてきているが、温室効果はフロン以上に大きく、また弗素が分解により環境中に放出されるなど、決して自由に放散が許される代替物ではない。したがって、回収、分解等の処置が望まれる。この報告では、OHラジカルを用いた代替フロンの超臨界水中での分解の素過程をHFC-32(2弗化メタン)を例に取り、非経験的分子軌道計算により解析した事例を述べている。分解反応全体としては、大きく発熱的であること、弗化水素の離脱では触媒として、超臨界水条件下で反応物に接した水分子が働くこと、この2点が重要知見だが、計算スキーム時には密度汎関数法の危険性も明らかになった。
望月 祐志; 田中 皓*
Theoretical Chemistry Accounts, 101(4), p.292 - 296, 1999/03
被引用回数:11 パーセンタイル:33.33(Chemistry, Physical)GaN固体は、青色領域以上の光を発する素材として極めて重要であり、その物性は実験、理論ともに精力的に研究されてきている。しかし、その単体-すなわちGaN分子そのもの-に関する研究はいまだ行われていない。本研究では、多参照の多電子理論である4次の結合電子対近似(MRCPA-4)を用い、GaN分子の分光学定数を基底状態、及び第一励起状態について高精度に評価した。MRCPA-4については、望月が作成する予定の分子ソフト:LCIにも組み込みを検討しており、本研究は``素材データ収集''の意味もある。
