


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
早川 頌*; 山本 耀二郎*; 沖田 泰良*; 板倉 充洋; 鈴木 克幸*
Computational Materials Science, 218, p.111987_1 - 111987_10, 2023/02
被引用回数:1 パーセンタイル:14.66(Materials Science, Multidisciplinary)On-the-fly kinetic Monte Carlo (kMC), a computational technique for atomistic simulations, has attracted attention because it increases the simulation timescale beyond that of molecular dynamics (MD) simulations while maintaining atomistic fidelity. However, for most kMC methods, when events with high and low activation energies coexist in the event list, trivial events with extremely low activation energies that do not essentially affect the phenomena of interest, so-called flicker events, are frequently selected, making it challenging to observe the key dynamics. In this study, we use Self-Evolving Atomistic kMC (SEAKMC), one of the on-the-fly kMC methods, to model the unstable-to-stable transformations of irregular three-dimensional self-interstitial-atom (SIA) clusters in Cu generated through collision cascade. By setting an activation energy threshold once every five steps, transformations into stable configurations are enhanced. The algorithm renders the simulation timescales one or two orders of magnitude longer than those possible with MD simulations. Further, the probability of transformations into stable configurations is increased by 40 times compared to that of the original SEAKMC method. In addition, we find that the stable configurations obtained by the transformation of the SIA clusters are mostly Frank loops. In summary, this new algorithm for the SEAKMC method helps to resolve the inefficiency of kMC methods resulting from the selection of flicker events and will aid the study of meso-timescale atomistic dynamics.
津川 聖人*; 早川 頌*; 沖田 泰良*; 愛知 正温*; 板倉 充洋; 鈴木 克幸*
Computational Materials Science, 215, p.111806_1 - 111806_8, 2022/12
被引用回数:2 パーセンタイル:29.01(Materials Science, Multidisciplinary)Molecular dynamics simulations were conducted to evaluate the interactions between an edge dislocation and a rigid, impenetrable precipitate in Cu by changing the distance between the glide plane of the dislocation and the center of the precipitate (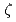 ). In these calculations, the precipitate was introduced as a super particle that moved according to the total force exerted by the matrix atoms on the precipitate atoms. When the center of the precipitate was close to the glide plane, an Orowan loop was formed around the precipitate after the dislocation detached, and the critical resolved shear stress (CRSS) was similar to the value evaluated by the results at
). In these calculations, the precipitate was introduced as a super particle that moved according to the total force exerted by the matrix atoms on the precipitate atoms. When the center of the precipitate was close to the glide plane, an Orowan loop was formed around the precipitate after the dislocation detached, and the critical resolved shear stress (CRSS) was similar to the value evaluated by the results at 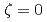 . However, when the glide plane was far from the center of the precipitate, either a vacancy loop or loops generated through the Hirsch mechanism were formed, depending on whether the center of the precipitate was below or above the glide plane. The magnitude of the CRSS was not symmetric about . This study confirmed that it is necessary to analyze the CRSS by changing to construct a predictive model for the hardening caused by the formation of lattice defects, and that precipitate hardening appears to be smaller than the value estimated using the results at .
. However, when the glide plane was far from the center of the precipitate, either a vacancy loop or loops generated through the Hirsch mechanism were formed, depending on whether the center of the precipitate was below or above the glide plane. The magnitude of the CRSS was not symmetric about . This study confirmed that it is necessary to analyze the CRSS by changing to construct a predictive model for the hardening caused by the formation of lattice defects, and that precipitate hardening appears to be smaller than the value estimated using the results at .
津川 聖人*; 早川 頌*; 岩瀬 祐樹*; 沖田 泰良*; 鈴木 克幸*; 板倉 充洋; 愛知 正温*
Computational Materials Science, 210, p.111450_1 - 111450_9, 2022/07
被引用回数:8 パーセンタイル:75.5(Materials Science, Multidisciplinary)Precipitation strengthening has been utilized to improve the properties of metallic materials so far. Since interactions between precipitates and dislocations are micro-mechanisms responsible for this phenomenon, a molecular dynamics (MD) simulation is a powerful tool for quantifying this phenomenon. In this study, we introduced a method to simulate a rigid and impenetrable precipitate against a direct contact with a dislocation using a single interatomic potential representing the bulk material. The total force exerted on all atoms in the precipitate region was divided by the number of atoms in the region. This average force was then applied to each atom in the region to simulate one super particle that moved depending on the total force exerted by the matrix atoms on the precipitate atoms. We used MD simulations to quantify the interaction of a precipitate with an edge dislocation. After the dislocation overcame the precipitate, an Orowan loop was formed along the outer circumference of the precipitate. The energy of the loop was 2.1 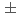 0.1 eV/b, which was higher than that obtained using the elasticity. The hardening caused by the precipitate was larger than that caused by voids of the same size. The proposed method can be applied to simulate interactions of precipitates with dislocations in any type of metallic material, especially when a dislocation bypasses a precipitate without changing its structure, except when a strong repulsive force acts between them.
0.1 eV/b, which was higher than that obtained using the elasticity. The hardening caused by the precipitate was larger than that caused by voids of the same size. The proposed method can be applied to simulate interactions of precipitates with dislocations in any type of metallic material, especially when a dislocation bypasses a precipitate without changing its structure, except when a strong repulsive force acts between them.
森 承宇*; 松田 那由多*; 沖田 泰良*; 愛知 正温*; 板倉 充洋; 鈴木 克幸*
Materialia, 21, p.101371_1 - 101371_6, 2022/03
The nonlinear ultrasonic (NLU) technique is a nondestructive method for detecting nanostructure in crystalline materials. In this study, a method was developed to quantify the changes in NLU signals associated with nanostructure using molecular dynamics (MD). A nonreflective boundary, which reduces the computational cost to the first power of the wavelength, was used to achieve this. This method is distinct from previous studies using a conventional MD, for which the computational cost is proportional to the square of the wavelength. The nonreflective boundary eliminates the influence of reflected waves at the detection position by setting a buffer region at the end of the simulation cell opposite from the wave source, and periodically resetting the displacements and velocities of all atoms in this region. This method allows the introduction of elastic waves with wavelengths longer than the cell size, and only an extension of time is required, according to the extension of the wavelength, without increasing the cell size. Hence, it is possible to extend the NLU wavelength by approximately four orders of magnitude, which approaches the wavelengths used for inspections and, thus, to use MD to simulate the changes in the NLU signals induced by nanostructure. The NLU signal values obtained by the two methods were in good agreement for a perfect Fe crystal and a Fe crystal containing 1% monovacancies. No significant frequency dependence of the acoustic nonlinearity parameter was found at 0 K. This method will contribute to the development of an inspection technique based on scientific principles.
沖田 泰良*; 寺山 怜志*; 津川 聖人*; 小林 恵太; 奥村 雅彦; 板倉 充洋; 鈴木 克幸*
Computational Materials Science, 202, p.110865_1 - 110865_9, 2022/02
被引用回数:7 パーセンタイル:48.44(Materials Science, Multidisciplinary)In this study, a Neural Network Potential (NNP) using an Artificial Neural Network (ANN) was developed for Zr, which is used as fuel cladding material in light water reactors. The reference data were obtained through first-principles calculations of various quantities, such as strained hexagonal-closed-packed (hcp) cells, strained face-centered cubic cells, cells containing a vacancy, several vacancies, and surfaces and 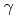 -surface energy on all five slip planes in the hcp structures. These data were converted to training data for the ANN, which were invariant to the rotation and translation of the atoms and independent of the number of atoms in the cells. The ANN was defined as a three-layer structure and the number of the nodes was set to 26-12-18-1. The NNP reproduced the first-principles calculations, particularly for the shear deformation, vacancy formation energy, surface energy, and -surface energy, with much higher accuracy than any of the existing potentials that have been developed for classical molecular dynamics simulations. The NNP was applied to identify the formation process of c-type dislocation loops in Zr, which is a key microstructure responsible for abrupt increases in hydrogen absorption. The formation process was determined by the balance of the vacancy formation energy, surface energy and the -surface energy on the basal plane, both of which were precisely reproduced only by the NNP developed in this study. The formation process was identified based on the atomistic behavior of the NNP.
-surface energy on all five slip planes in the hcp structures. These data were converted to training data for the ANN, which were invariant to the rotation and translation of the atoms and independent of the number of atoms in the cells. The ANN was defined as a three-layer structure and the number of the nodes was set to 26-12-18-1. The NNP reproduced the first-principles calculations, particularly for the shear deformation, vacancy formation energy, surface energy, and -surface energy, with much higher accuracy than any of the existing potentials that have been developed for classical molecular dynamics simulations. The NNP was applied to identify the formation process of c-type dislocation loops in Zr, which is a key microstructure responsible for abrupt increases in hydrogen absorption. The formation process was determined by the balance of the vacancy formation energy, surface energy and the -surface energy on the basal plane, both of which were precisely reproduced only by the NNP developed in this study. The formation process was identified based on the atomistic behavior of the NNP.
寺山 怜志*; 岩瀬 祐樹*; 早川 頌*; 沖田 泰良*; 板倉 充洋; 鈴木 克幸*
Computational Materials Science, 195, p.110479_1 - 110479_12, 2021/07
被引用回数:9 パーセンタイル:57.69(Materials Science, Multidisciplinary)Austenitic stainless steels, which are used as incore structural materials in light water reactors, are characterized by an extremely low stacking fault energy (SFE) among face-centered cubic (FCC) metals. To evaluate the effects of SFE on defect formation under high-energy particle irradiation, molecular dynamics simulations were performed using the interatomic potential sets for FCC metals with different SFEs and a primary knock-on atom energy (E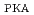 ) of 100 keV at 600 K. The results show that the number of residual defects is independent of the SFE. However, the characteristics of self-interstitial atom (SIA) clusters do depend on the SFE. For clusters smaller than a certain size, the ratio of glissile SIA clusters decreases as the SFE increases, which is similar to the trend observed at the low E. However, for larger clusters, which can be detected only at a high E, the ratio of glissile clusters increases. These results correspond to static energy calculations, in which the difference in the formation energy between a Frank loop and perfect loop (
) of 100 keV at 600 K. The results show that the number of residual defects is independent of the SFE. However, the characteristics of self-interstitial atom (SIA) clusters do depend on the SFE. For clusters smaller than a certain size, the ratio of glissile SIA clusters decreases as the SFE increases, which is similar to the trend observed at the low E. However, for larger clusters, which can be detected only at a high E, the ratio of glissile clusters increases. These results correspond to static energy calculations, in which the difference in the formation energy between a Frank loop and perfect loop (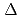 E
E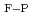 ) for the small clusters decreases as the SFE increases. In contrast, for the larger clusters, the SFE dependence of E changes due to the shape restrictions of stable perfect loops. At a high temperature of 600 K, large vacancy clusters with stacking faults can be detected at E = 100 keV, resulting in the enhanced formation of these clusters at lower SFEs. Furthermore, several of these clusters were similar to perfect loops, with the edges split into two partial dislocations with stacking faults, although the largest clusters detected at low Es were similar to stacking fault tetrahedrons.
) for the small clusters decreases as the SFE increases. In contrast, for the larger clusters, the SFE dependence of E changes due to the shape restrictions of stable perfect loops. At a high temperature of 600 K, large vacancy clusters with stacking faults can be detected at E = 100 keV, resulting in the enhanced formation of these clusters at lower SFEs. Furthermore, several of these clusters were similar to perfect loops, with the edges split into two partial dislocations with stacking faults, although the largest clusters detected at low Es were similar to stacking fault tetrahedrons.
早川 頌*; 土井原 康平*; 沖田 泰良*; 板倉 充洋; 愛知 正温*; 鈴木 克幸*
Journal of Materials Science, 54(17), p.11509 - 11525, 2019/09
被引用回数:15 パーセンタイル:56.33(Materials Science, Multidisciplinary)We performed molecular dynamics simulations to evaluate the effects of stacking fault energy (SFE) on interactions between a screw dislocation and spherical voids in face-centered cubic (fcc) metals. It was observed that the frequency of the cross-slips is a critical factor affecting the interaction, with primarily three different interaction morphologies being observed: (1) the two partial dislocations detach from the void independently with a time lag, (2) the two partial dislocations detach from the void almost simultaneously on a single slip plane, and (3) the two partial dislocations detach from the void almost simultaneously while involving more than one cross-slip and a jog formation. The magnitude of the critical resolved shear stress (CRSS) increases in the order mentioned above. The CRSS values for interaction morphology (2), which was observed most frequently in this study, were in good agreement with those predicted analytically by adjusting the parameters dependent on the SFE. Based on the obtained results, we discussed the applicability of the analytical model for void hardening in fcc metals. The results of this work contribute significantly to the modeling of mechanical property degradation in irradiated metals.
早川 頌*; 沖田 泰良*; 板倉 充洋; 川畑 友弥*; 鈴木 克幸*
Journal of Materials Science, 54(16), p.11096 - 11110, 2019/08
被引用回数:11 パーセンタイル:42.21(Materials Science, Multidisciplinary)We performed molecular dynamics simulations of displacement cascades in FCC metals under Poisson's deformation using interatomic potentials differing in stacking fault energy (SFE), in order to investigate the effect of tensile strain on the SFE dependence of defect formation processes. There was no clear SFE dependence of the number of residual defects and the size distribution of defect clusters under both no strain and the applied strain, while the strain enhanced the defect formation to a certain extent. We also observed that the strain affected the formations of self-interstitial atom (SIA) clusters depending on their size and the Burgers vector. These results were consistent with the analysis based on the defect formation energies. Meanwhile, the number of SIA perfect loops was higher at lower SFE under both no strain and the applied strain, leading to an increase in the ratio of glissile SIA clusters with a decrease in SFE. Further, the absolute number of SIA perfect loops was increased by the applied strain, while the SFE dependence of the number of SIA perfect loops was not affected. These findings were associated with the difference in formation energy between an SIA perfect loop and an SIA Frank loop. The insights extracted from this study significantly contribute to the modeling of microstructural evolution in nuclear materials under irradiation, especially for low SFE metals such as austenitic stainless steels.
中西 大貴*; 川畑 友弥*; 土井原 康平*; 沖田 泰良*; 板倉 充洋; 鈴木 克幸*
Philosophical Magazine, 98(33), p.3034 - 3047, 2018/09
被引用回数:10 パーセンタイル:47.84(Materials Science, Multidisciplinary)原子炉内部で使用されるオーステナイト鋼は積層欠陥エネルギーが低いことで知られ、そのため積層欠陥を伴う照射欠陥クラスタが大きくなることが予想される。この傾向を定量評価するため、FCC金属のモデルポテンシャルにおいて積層欠陥エネルギーだけを変化させたものを用い照射欠陥生成の分子動力学シミュレーションを様々な温度において実行し、生成した照射欠陥クラスタの種類と数を調べた。その結果移動可能な格子間原子クラスタの生成数が積層欠陥エネルギーとともに大きく変化することが見出された。
早川 頌*; 沖田 泰良*; 板倉 充洋; 愛知 正温*; 鈴木 克幸*
Philosophical Magazine, 98(25), p.2311 - 2325, 2018/06
被引用回数:8 パーセンタイル:40.77(Materials Science, Multidisciplinary)BCC鉄における格子間原子クラスタは容易方向へのすべり運動とは別に垂直方向への保存的上昇運動を行い、これにより三次元的な運動を行う。この拡散の三次元性は転位への吸収確率に大きく影響し、材料のスエリングなどの照射劣化挙動を左右する。この保存的上昇運動を分子動力学計算とモンテカルロ計算を組み合わせた手法で解析し、移動障壁エネルギーを評価したところ、従来の弾性理論により予想されていた値よりはるかに大きい値が得られた。
土井原 康平*; 沖田 泰良*; 板倉 充洋; 愛知 正温*; 鈴木 克幸*
Philosophical Magazine, 98(22), p.2061 - 2076, 2018/05
被引用回数:20 パーセンタイル:71.42(Materials Science, Multidisciplinary)原子炉材料、特に炉内構造材のオーステナイトの照射脆化を評価する際に、オーステナイトの特徴である低い積層欠陥エネルギーの影響を評価する必要がある。本研究では積層欠陥エネルギーだけを変化させたFCC金属のモデルポテンシャルを用いて転位とボイドの相互作用形態を評価し、ボイドの大きさと積層欠陥エネルギーの値によって様々な相互作用形態があることを見出した。
沖田 泰良*; 板倉 充洋
日本原子力学会誌ATOMO , 59(12), p.712 - 716, 2017/12
, 59(12), p.712 - 716, 2017/12
原子力材料の照射下特性変化を原子スケールの現象に基づいてモデル化する分子シミュレーションは、極短時間・極微小スケールの現象を定量化・解明・再現するため、幅広く用いられるようになってきた。本稿では、(1)分子動力学法を用いた高速中性子との弾性衝突に伴う結晶欠陥形成過程の定量化、(2)分子動力学法を用いた結晶欠陥集合体-転位接触による転位チャネリング形成過程の解明、(3)分子動力学-キネッティックモンテカルロ融合法を用いた結晶欠陥集合体の3次元的運動・転位へ吸収される過程の再現、に関して近年の研究進展を紹介する。
浅利 圭亮*; Hetland, O. S.*; 藤田 智*; 板倉 充洋; 沖田 泰良*
Journal of Nuclear Materials, 442(1), p.360 - 364, 2013/11
被引用回数:21 パーセンタイル:83.85(Materials Science, Multidisciplinary)炉内構造材料であるオーステナイトについて、その特徴である低い積層欠陥エネルギーが照射脆化にどう影響するかを調べるため、積層欠陥エネルギーの異なる分子動力学ポテンシャルを用いて照射欠陥による脆化への影響を調べた。その結果、積層欠陥エネルギーが低い場合は転位が広く分裂し二本の独立な転位として照射欠陥から抜けていく過程が観察され、またその過程に必要な外部応力が低下することが分かった。この結果はオーステナイトにおける脆化のモデル化に必須な情報となる。
板倉 充洋; 蕪木 英雄; 山口 正剛; 沖田 泰良*
Acta Materialia, 61(18), p.6857 - 6867, 2013/10
被引用回数:115 パーセンタイル:98.16(Materials Science, Multidisciplinary)原子力材料は長年の中性子照射によって硬化する。これは金属材料が折れることなく曲がる塑性変形が、照射による材料変化によって阻害される現象であるが、これを解明するには塑性のメカニズムを原子スケールで明らかにする必要がある。それには塑性変形を担う転位線と呼ばれる格子欠陥の動きがどのように照射損傷により阻害されるを知る必要があり、これは実験で直接観察できないので大規模な量子計算が必要になる。本発表では量子計算によって初めてこの転位の移動阻害メカニズムを明らかにしたので報告する。大規模な計算が必要という課題の解決にあたっては、新たに考案した多階層計算手法を用い少ない原子数で多数の原子での計算に相当する精度を出すことを可能にしたことが上げられる。これによって転位の運動阻害プロセスを定量的にモデル化することが可能となり、照射硬化をシミュレーションで定量評価するための道が開けたと言える。
沖田 泰良*; 越石 正人*; 橋本 素行*; 田中 重彰*; 児玉 光弘*; 近藤 啓悦; 塚田 隆
no journal, ,
経済産業省原子力安全・保安院高経年化対策基盤整備事業の一環として、軽水炉炉内構造材であるオーステナイトステンレス鋼の照射誘起応力腐食割れのき裂進展挙動に及ぼす照射速度の影響について検討を行った。本研究では、き裂進展速度データが既に取得されている実機材を用いて、照射硬化,ひずみ硬化指数,照射欠陥及び粒界偏析に関するデータを取得し、それらデータから既存のき裂進展モデル(FRIモデル)への入力パラメータを決定してき裂進展速度を計算し、実験値との比較を行った。その結果、照射硬化や照射誘起偏析に伴う耐食性の劣化をモデル計算にとり入れることによって、き裂進展速度実験データを説明できることを実証した。これらの検討の結果、照射の影響を反映したパラメータ選定によって、FRIモデルを照射材き裂進展計算に適用可能であることがわかった。
板倉 充洋; 宮代 聡*; 沖田 泰良*
no journal, ,
現在さまざまな金属について性能の良い分子動力学ポテンシャルが開発され、大規模な分子動力学計算によって新たな知見が得られているが、実用材料、特に合金に関しては分子動力学ポテンシャルの開発はあまり進んでいない。オーステナイト合金の軽水炉での照射脆化等の計算科学研究によるモデル化には分子動力学ポテンシャルの開発が必須であるため、その出発点として本研究では基本的な物質特性を再現するポテンシャルを開発して基礎プラットフォームを構築する。これまでの研究で、鉄・クロム・ニッケルをランダムに配置した小規模な系での電子構造計算を多数のケースについて行い、このような三元系のポテンシャル開発を行う場合に必要となる電子構造計算の原子配置ケース数や計算精度を見積もった。その結果、32原子程度の小規模な系では50ケース程度の原子配置ケースが必要であること、及び各原子の最近接ペア間の相互作用は最大でも0.08eV程度と大きくなく、オーステナイト鋼が急冷されて製造されることを考慮すると、電子構造計算において完全にランダムな配置を用いることは物理的にも妥当であることがわかった。今後このポテンシャルを改良し点欠陥や点欠陥クラスタのエネルギー及び移動障壁エネルギーが再現できるようになれば、照射によって生成する格子欠陥の分布を知ることが可能となり、また耐蝕性を担うクロム原子が照射により材料中でどのように移動するかを調べることで照射が耐蝕性に与える影響を調べることが可能となる。
板倉 充洋; 宮代 聡*; 沖田 泰良*; 山口 正剛; 奥田 洋司*
no journal, ,
本研究テーマの最終目的は、主要な原子炉構造材料の一つである鉄・クロム・ニッケル合金について、原子スケールでの経年劣化シミュレーションのために必須となる原子間ポテンシャルを開発することである。そのためにはさまざまな原子配置について第一原理計算を行い、その結果をよく再現するポテンシャル関数を作成することが必要となるが、合金の場合は原子配置の数が組合せ論的に膨大となる困難が存在する。この課題を解決するため、まずは小規模な系において60ケースのランダムな原子配置を作成して第一原理計算を行い、配置の違いによってどの程度エネルギーなどの物理量が変化するかを調べた。その結果、鉄・クロム・ニッケル合金の場合は元素の違いによる原子間相互作用の変化が小さく、比較的少数のケース数に基づいた計算でも十分信頼できるデータを得られることがわかった。この結果により、現在の計算機の能力でこの合金のポテンシャル開発に必要な第一原理計算を行うことは十分可能であることがわかり、実用原子炉材料の経年劣化シミュレーション実現へ向けて大きく前進することができた。
板倉 充洋; 沖田 泰良*
no journal, ,
現在核廃棄物を減らす新型原子炉の設計が行われており、その原子炉で使用する燃料被覆管の安全性について原子力機構, 東京大学, 日立製作所で共同研究を行っている。これまでより強い照射条件において被覆管の寿命の決定因子である水素化物析出までの時間がどのように変化するかを理論的なモデルにより予測し、適切な材料を開発することが目的である。本発表では照射欠陥クラスタへの水素偏析が駆動現象になると仮定し、その仮定を検証するため第一原理計算で純Zr中の空孔クラスタや積層欠陥への偏析エネルギー等を評価した結果を公表する。得られた結果は、最終的な材料開発に不可欠なモデルのパラメータを与えるものであり、新型炉の設計に大きく貢献するものである。
板倉 充洋; 沖田 泰良*
no journal, ,
現在核廃棄物を減らす新型原子炉の設計が行われており、その原子炉で使用する燃料被覆管の安全性について原子力機構, 東京大学, 日立製作所で共同研究を行っている。これまでより強い照射条件において被覆管の寿命の決定因子である水素化物析出までの時間がどのように変化するかを理論的なモデルにより予測し、適切な材料を開発することが目的である。本発表では照射効果のモデル化のため、照射試料において顕著である鉄クロム析出物の溶出と水素の関係を明らかにするため、特に鉄原子が照射にどのような影響を受けるかを第一原理計算で調べた結果を公表する。得られた結果は、最終的な材料開発に不可欠なモデルのパラメータを与えるものであり、新型炉の設計に大きく貢献するものである。
板倉 充洋; 沖田 泰良*
no journal, ,
現在核廃棄物を減らす新型原子炉の設計が行われており、その原子炉で使用する燃料被覆管の安全性について原子力機構, 東京大学, 日立製作所で共同研究を行っている。これまでより強い照射条件において被覆管の寿命の決定因子である水素化物析出までの時間がどのように変化するかを理論的なモデルにより予測し、適切な材料を開発することが目的である。本発表では照射欠陥クラスタへの水素偏析が駆動現象になると仮定し、その仮定を検証するため第一原理計算で純Zr中の空孔クラスタや積層欠陥への偏析エネルギー等を評価した結果を公表する。得られた結果は、最終的な材料開発に不可欠なモデルのパラメータを与えるものであり、新型炉の設計に大きく貢献するものである。
