


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Akutsu, Kazuhiro*; Adachi, Motoyasu*; Kawakita, Yukinobu
JAEA-Review 2018-002, 36 Pages, 2018/03
 JAEA-Review-2018-002.pdf:4.67MB
JAEA-Review-2018-002.pdf:4.67MB
Since J-PARC started operation for user programs with high-intensity pulsed neutron beams, many material and life science studies have been carried out at J-PARC. Since partial or complete deuteration of organic compounds for contrast variation in the scattering length densities of materials is one of the most effective techniques in the application of neutron scattering analysis, deuterated materials have led to significant progress in our understanding of the structure of novel organic materials. Aiming at accelerating development of deuteration activities in our country, an international workshop "Deuterated Materials Enhancing Neutron Science for Structure Function Applications" was held as a J-PARC Workshop at Ibaraki Quantum Beam Research Center from 19th October to 20th October in 2017. In this workshop, the chemical/biological deuteration activities and recent scientific results of deuterated materials enhancing neutron study in Japan and other countries were discussed by domestic/foreign deuteration and neutron scientists. This is a report of the workshop summarized by organizers.
 binding site of a
binding site of a 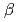 -lactamase from the halophile by anomalous X-ray diffraction
-lactamase from the halophile by anomalous X-ray diffractionArai, Shigeki; Shibazaki, Chie; Shimizu, Rumi; Adachi, Motoyasu; Tamada, Taro; Tokunaga, Hiroko*; Ishibashi, Matsujiro*; Tokunaga, Masao*; Kuroki, Ryota
Kyushu Shinkurotoronko Kenkyu Senta Nempo, 2014, p.17 - 19, 2016/03
no abstracts in English
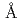 resolution
resolutionUemura, Takuya*; Kita, Akiko*; Watanabe, Yoshihiko*; Adachi, Motoyasu; Kuroki, Ryota; Morimoto, Yukio*
Biochemical and Biophysical Research Communications, 469(2), p.158 - 163, 2016/01
Times Cited Count:18 Percentile:53.10(Biochemistry & Molecular Biology)Hiromoto, Takeshi; Adachi, Motoyasu; Shibazaki, Chie; Schrader, T. E.*; Ostermann, A.*; Kuroki, Ryota
JPS Conference Proceedings (Internet), 8, p.033003_1 - 033003_6, 2015/09
T4 phage lysozyme (T4L) is an endoacetylmuramidase that degrades the murein of the bacterial cell wall by cleaving the -1,4-glycosidic bond between 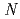 -acetylmuramic acid and -acetylglucosamine. We previously reported that the substitution of the catalytic Thr26 with the nucleophilic His converts the wild-type (WT) T4L from an inverting to a retaining glycosidase, in which the -configuration of the substrate is retained in the product. We also found that the Thr26His (T26H) mutant can catalyze a transglycosylation reaction more effectively than hydrolysis, although the WT-T4L has no transglycosidase activity. To clarify the role of the substituted His26 in transglycosylation and to investigate its relationship to the neighboring acidic residue Asp20 using neutron crystallography, a perdeuterated recombinant protein of the T26H mutant (d-T26H) was prepared for crystallization. The perdeuterated form was produced in
-acetylmuramic acid and -acetylglucosamine. We previously reported that the substitution of the catalytic Thr26 with the nucleophilic His converts the wild-type (WT) T4L from an inverting to a retaining glycosidase, in which the -configuration of the substrate is retained in the product. We also found that the Thr26His (T26H) mutant can catalyze a transglycosylation reaction more effectively than hydrolysis, although the WT-T4L has no transglycosidase activity. To clarify the role of the substituted His26 in transglycosylation and to investigate its relationship to the neighboring acidic residue Asp20 using neutron crystallography, a perdeuterated recombinant protein of the T26H mutant (d-T26H) was prepared for crystallization. The perdeuterated form was produced in 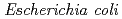 cells cultured in deuterated-rich media. After purification, the d-T26H mutant was crystallized under deuterated conditions and grown to a volume of 0.12 mm
cells cultured in deuterated-rich media. After purification, the d-T26H mutant was crystallized under deuterated conditions and grown to a volume of 0.12 mm using a macroseeding technique. A preliminary neutron-diffraction experiment at 100 K at the FRM II research reactor (Munich, Germany) gave diffraction spots of up to 2.8 resolution after a 1.5 h exposure.
using a macroseeding technique. A preliminary neutron-diffraction experiment at 100 K at the FRM II research reactor (Munich, Germany) gave diffraction spots of up to 2.8 resolution after a 1.5 h exposure.
-lactamase from the moderate halophile 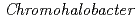 sp.560 and the discovery of a Cs-selective binding site
sp.560 and the discovery of a Cs-selective binding siteArai, Shigeki; Yonezawa, Yasushi*; Okazaki, Nobuo*; Matsumoto, Fumiko*; Shibazaki, Chie; Shimizu, Rumi; Yamada, Mitsugu*; Adachi, Motoyasu; Tamada, Taro; Kawamoto, Masahide*; et al.
Acta Crystallographica Section D, 71(3), p.541 - 554, 2015/03
Times Cited Count:8 Percentile:52.61(Biochemical Research Methods)The crystal structure of halophilic -lactamase from sp.560 (HaBLA) was determined using X-ray crystallography. Moreover, the locations of bound Sr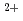 and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-
and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-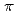 interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
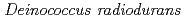
Adachi, Motoyasu; Hirayama, Hiroshi; Shimizu, Rumi; Sato, Katsuya; Narumi, Issey*; Kuroki, Ryota
Protein Science, 23(10), p.1349 - 1358, 2014/10
Times Cited Count:10 Percentile:26.31(Biochemistry & Molecular Biology)Pleiotropic protein promoting DNA repair A (PprA) is a key protein that facilitates the extreme radioresistance of . To clarify the role of PprA in the radioresistance mechanism, the interaction between recombinant PprA expressed in Escherichia coli with several double-stranded DNAs was investigated. In a gel-shift assay, the band shift of supercoiled pUC19 DNA caused by the binding of PprA showed a bimodal distribution, which was promoted by the addition of 1 mM Mg, Ca, or Sr ions. The dissociation constant of the PprA-supercoiled pUC19 DNA complex, calculated from the relative portions of shifted bands, was 0.6 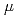 M with a Hill coefficient of 3.3 in the presence of 1 mM Mg acetate. This indicates that at least 281 PprA molecules are required to saturate a supercoiled pUC19 DNA, which is consistent with the number of bound PprA molecules estimated by the UV absorption of the PprA-pUC19 complex purified by gel filtration. This saturation also suggests linear polymerization of PprA along the dsDNA. On the other hand, the bands of linear dsDNA and nicked circular dsDNA that eventually formed PprA complexes did not saturate, but created larger molecular complexes when the PprA concentration was greater than 1.3 M. This result implies that DNA-bound PprA aids association of the termini of damaged DNAs, which is regulated by the concentration of PprA.
M with a Hill coefficient of 3.3 in the presence of 1 mM Mg acetate. This indicates that at least 281 PprA molecules are required to saturate a supercoiled pUC19 DNA, which is consistent with the number of bound PprA molecules estimated by the UV absorption of the PprA-pUC19 complex purified by gel filtration. This saturation also suggests linear polymerization of PprA along the dsDNA. On the other hand, the bands of linear dsDNA and nicked circular dsDNA that eventually formed PprA complexes did not saturate, but created larger molecular complexes when the PprA concentration was greater than 1.3 M. This result implies that DNA-bound PprA aids association of the termini of damaged DNAs, which is regulated by the concentration of PprA.
 sp.593
sp.593Arai, Shigeki; Yonezawa, Yasushi*; Ishibashi, Matsujiro*; Matsumoto, Fumiko*; Adachi, Motoyasu; Tamada, Taro; Tokunaga, Hiroko*; Blaber, M.; Tokunaga, Masao*; Kuroki, Ryota
Acta Crystallographica Section D, 70(3), p.811 - 820, 2014/03
Times Cited Count:13 Percentile:63.53(Biochemical Research Methods)In order to clarify the structural basis of halophilic characteristics of an alkaline phosphatase derived from the moderate halophile sp.593 (HaAP), the tertiary structure of HaAP was determined to 2.1 resolution by X-ray crystallography. Structural properties of surface negative charge and core hydrophobicity are shown to be intermediate between halophile and non-halophile characteristics, and may explain the unique functional adaptation to a wide-range of salt concentration.
Adachi, Motoyasu; Arai, Shigeki; Hiromoto, Takeshi; Kuroki, Ryota
Hamon, 24(1), p.45 - 49, 2014/02
Protein structure analysis using neutron diffraction (neutron protein crystallography; NPC) is gaining greater importance in the understanding of structure and function relationships of biological macromolecules such as proteins and DNA. Current developments of neutron diffractometers installed at the JAEA research reactor and pulsed neutron source permit observation of the locations of hydrogen atoms and hydrating water molecules and help understanding of important mechanisms of chemical reactions catalyzed by biological macromolecules. Here, we introduce practical approaches of NPC including sample preparation, crystal growth, structure determination and utilization of information obtained from NPC.
Adachi, Motoyasu; Shimizu, Rumi; Kuroki, Ryota; Blaber, M.
Journal of Synchrotron Radiation, 20(6), p.953 - 957, 2013/11
Times Cited Count:2 Percentile:12.54(Instruments & Instrumentation)Symfoil-4P is a 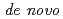 protein exhibiting the threefold symmetrical beta-trefoil fold designed based on the human acidic fibroblast growth factor. First three asparagine-glycine sequences of Symfoil-4P are replaced with glutamine-glycine (Symfoil-QG) or serine-glycine (Symfoil-SG) sequences protecting from deamidation, and His-Symfoil-II was prepared by introducing a protease digestion site into Symfoil-QG so that Symfoil-II has three complete repeats after removal of the N-terminal histidine tag. The Symfoil-QG and SG and His-Symfoil-II proteins were expressed in
protein exhibiting the threefold symmetrical beta-trefoil fold designed based on the human acidic fibroblast growth factor. First three asparagine-glycine sequences of Symfoil-4P are replaced with glutamine-glycine (Symfoil-QG) or serine-glycine (Symfoil-SG) sequences protecting from deamidation, and His-Symfoil-II was prepared by introducing a protease digestion site into Symfoil-QG so that Symfoil-II has three complete repeats after removal of the N-terminal histidine tag. The Symfoil-QG and SG and His-Symfoil-II proteins were expressed in 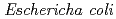 as soluble protein, and purified by nickel affinity chromatography. Symfoil-II was further purified by anion-exchange chromatography after removing the HisTag by proteolysis. Symfoil-QG and II crystals gave 1.5 and 1.1, resolution, respectively. The refined crystal structure of Symfoil-II showed pseudo-threefold symmetry as expected from other Symfoils.
as soluble protein, and purified by nickel affinity chromatography. Symfoil-II was further purified by anion-exchange chromatography after removing the HisTag by proteolysis. Symfoil-QG and II crystals gave 1.5 and 1.1, resolution, respectively. The refined crystal structure of Symfoil-II showed pseudo-threefold symmetry as expected from other Symfoils.
Tamada, Taro; Adachi, Motoyasu; Kurihara, Kazuo; Kuroki, Ryota
Nihon Kessho Gakkai-Shi, 55(1), p.47 - 51, 2013/02
Neutron crystallography enables us to identify the accurate hydrogen positions in proteins, which play important roles in many chemical reactions in living system. Here we show our results of neutron structure determination of enzymes in complex with its inhibitors which corresponds to transition state analogues. Neutron structure analysis elucidated the detail catalytic reaction of each enzyme by direct observation of hydrogen atoms. Furthermore we would like to introduce a new neutron beam line for neutron structural biology planned at MLF in J-PARC.
Nakaniwa, Tetsuko*; Fukata, Harumi*; Inoue, Tatsuya*; Goda, Masaki*; Nakai, Ryoko*; Kirii, Yasuyuki*; Adachi, Motoyasu; Tamada, Taro; Segawa, Shinichi*; Kuroki, Ryota; et al.
Biochemistry, 51(42), p.8410 - 8421, 2012/10
Times Cited Count:20 Percentile:44.63(Biochemistry & Molecular Biology)Protein kinase is a vital drug target for the treatment of a wide range of diseases. To investigate the effect of cysteine mutation on the function, stability and structure of kinase, free cysteines of c-Jun N-terminal kinase 1 (JNK1) were systematically removed by mutation. Two cysteine-destructed mutants in which three (M3) and seven (M7) cysteine residues are removed, yielded about 5 and 2 times than wild type JNK-1 (M0). SDS PAGE analysis showed that the aggregation was less in the case of M3 and M7. Thermal unfolding experiment of M0, M3 and M7 using by differential scanning calorimetry proceeded at least three state unfolding. Crystal structure of the M3 mutant was determined to 2.6 resolution, which was identical to that of the wild-type. Consequently, due to the highest yield, its improved stability against aggregation and its structural similarity to the wild type, the M3 mutant is suitable for the use of further characterization of its function and structure.
 HB8
HB8Okazaki, Nobuo; Adachi, Motoyasu; Tamada, Taro; Kurihara, Kazuo; Oga, Takushi*; Kamiya, Nobuo*; Kuramitsu, Seiki*; Kuroki, Ryota
Acta Crystallographica Section F, 68(1), p.49 - 52, 2012/01
Times Cited Count:2 Percentile:31.80(Biochemical Research Methods)
Kosaka, Nami*; Sugai, Tatsuhisa*; Nagasawa, Kazumichi*; Tanizaki, Yuta*; Meguro, Mizue; Aizawa, Yoichi*; Maekawa, Shun*; Adachi, Motoyasu; Kuroki, Ryota; Kato, Takashi
Journal of Experimental Biology, 214(6), p.921 - 927, 2011/03
Times Cited Count:32 Percentile:81.47(Biology)Oxygen is essential for the survival of animals. Red blood cells are responsible for transporting oxygen to tissues. We established a semi-solid colony forming assay, and showed that recombinant xlEPO induces erythroid colony formation in vitro and detected an increased level of erythropoietin activity in blood serum during acute anemic stress. In addition, the study demonstrated the possible presence of multiple, non-xlEPO, factors in anemic serum supportive of erythroid colony formation. These results indicate that erythropoiesis mediated by erythropoietin is present in amphibian species and, furthermore, that the regulatory mechanisms controlling peripheral erythrocyte number may vary among vertebrates.
Kuroki, Ryota; Okazaki, Nobuo; Adachi, Motoyasu; Ohara, Takashi; Kurihara, Kazuo; Tamada, Taro
Acta Crystallographica Section D, 66(11), p.1126 - 1130, 2010/11
Times Cited Count:2 Percentile:29.26(Biochemical Research Methods)It is generally known that enzymes represent important drug-target proteins. Elucidation of the catalytic function and the molecular-recognition mechanisms of enzymes provides important information for structure-based drug design. Neutron crystallography provides accurate information on the locations of H atoms that are essential in enzymatic function and molecular recognition. Recent examples are described of the structure determination of the drug-target proteins human immunodeficiency virus protease and porcine pancreatic elastase in complex with transition-state analogue inhibitors using the neutron diffractometers for biological crystallography (BIX-3 and BIX-4) installed at the JRR-3 research reactor.
resolutionShimizu, Noriko*; Sugiyama, Shigeru*; Maruyama, Mihoko*; Takahashi, Yoshinori*; Adachi, Motoyasu; Tamada, Taro; Hidaka, Koshi*; Hayashi, Yoshio*; Kimura, Toru*; Kiso, Yoshiaki*; et al.
Crystal Growth & Design, 10(7), p.2990 - 2994, 2010/06
Times Cited Count:11 Percentile:71.28(Chemistry, Multidisciplinary)We report crystal growth of human immunodeficiency virus 1 protease (HIV PR) in a complex with its inhibitor KNI-272 by six different methods. Comparative analysis indicates that top-seeded solution growth (TSSG) and TSSG combined with the floating and stirring technique (TSSG-FAST) are efficient strategies for rapidly obtaining large single crystals and effectively preventing polycrystallization of the seed crystal. Neutron diffraction analysis confirmed that the crystalobtained by TSSG is a high-quality single crystal. Furthermore, crystal shape was observed to be influenced by solution flow, suggesting that the degree of supersaturation significantly affects the crystal growth direction of HIV PR complex. This finding implies that the shape of the HIV PR complex crystal might be controlled by the solution flow rate.
Kuroki, Ryota; Tamada, Taro; Kurihara, Kazuo; Ohara, Takashi; Adachi, Motoyasu
Yakugaku Zasshi, 130(5), p.657 - 664, 2010/05
Times Cited Count:0 Percentile:0.00(Pharmacology & Pharmacy)Crystallography enables us to obtain accurate atomic positions within proteins. High resolution X-ray crystallography provides information for most of the atoms comprising a protein, with the exception of hydrogens. Neutron diffraction data can provide information of the location of hydrogen atoms to the structural information determined by X-ray crystallography. Here, we show the recent result of the structural determination of drug-target proteins, porcine pancreatic elastase and human immuno-deficiency virus type-1 protease by both X-ray and neutron diffraction. The structure of porcine pancreatic elastase with its potent inhibitor was determined at room temperature to 1.2 resolution by X-ray diffraction and 1.65 resolution by neutron diffraction. The structure of HIV-PR with its potent inhibitor was also determined to 1.4 resolution by X-ray diffraction and 1.9 resolution by neutron diffraction. Ultra-high resolution structures of both proteins (0.94 and 0.93 , respectively) were also determined by X-ray diffraction at 100 K. The ionization state and the location of hydrogen atoms of the catalytic residue in these enzymes were determined by neutron diffraction. Furthermore, collaborative use of both X-ray and neutron to identify the location of ambiguous hydrogen atoms will be shown.
Adachi, Motoyasu; Sunami, Tomoko; Kuroki, Ryota
Koso Riyo Gijutsu Taikei, p.34 - 37, 2010/04
no abstracts in English
Tamada, Taro; Adachi, Motoyasu
Radioisotopes, 59(4), p.299 - 308, 2010/04
Neutron diffraction data can provide information of the location of hydrogen atoms to the structural information determined by X-ray crystallography. Here, we show the recent results of the structural determination of drug-target proteins, porcine pancreatic elastase and human immuno-deficiency virus type-1 protease by both X-ray and neutron diffraction. The structure of porcine pancreatic elastase with its potent inhibitor was determined to 0.094 nm resolution by X-ray diffraction and 0.165 nm resolution by neutron diffraction. The structure of HIV-PR with its potent inhibitor was also determined to 0.093 nm resolution by X-ray diffraction and 0.19nm resolution by neutron diffraction. The ionization state and the location of hydrogen atoms of the catalytic residue in these enzymes were determined by neutron diffraction.
Adachi, Motoyasu; Kuroki, Ryota
Tampakushitsu Kakusan Koso, 55(1), p.82 - 87, 2009/12
no abstracts in English
Adachi, Motoyasu; Kuroki, Ryota
Hamon, 19(4), p.214 - 217, 2009/10
To develop HIV-1 protease inhibitors through structure-based drug design, it is necessary to understand the catalytic mechanism and inhibitor recognition of HIV-1 protease. We have determined the crystal structure of HIV-1 protease in complex with KNI-272 to 1.9 resolution by neutron crystallography in combination with 1.4 resolution X-ray diffraction data. The results show that the carbonyl group of hydroxymethylcarbonyl (HMC) in KNI-272 forms a hydrogen bonding interaction with protonated Asp 25 and the hydrogen atom from the hydroxyl group of HMC forms a hydrogen bonding interaction with the deprotonated Asp125. This is the first neutron report for HIV-1/inhibitor complex and shows directly the locations of key hydrogen atoms in catalysis and in the binding of a transition-state analog.
