


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
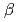 -lactamase from the moderate halophile
-lactamase from the moderate halophile 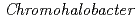 sp.560 and the discovery of a Cs
sp.560 and the discovery of a Cs -selective binding site
-selective binding siteArai, Shigeki; Yonezawa, Yasushi*; Okazaki, Nobuo*; Matsumoto, Fumiko*; Shibazaki, Chie; Shimizu, Rumi; Yamada, Mitsugu*; Adachi, Motoyasu; Tamada, Taro; Kawamoto, Masahide*; et al.
Acta Crystallographica Section D, 71(3), p.541 - 554, 2015/03
Times Cited Count:8 Percentile:52.42(Biochemical Research Methods)The crystal structure of halophilic -lactamase from sp.560 (HaBLA) was determined using X-ray crystallography. Moreover, the locations of bound Sr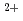 and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-
and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-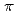 interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
 -glucosyltransferase from
-glucosyltransferase from 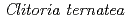
Hiromoto, Takeshi; Honjo, Eijiro*; Noda, Hisanobu*; Tamada, Taro; Kazuma, Kohei*; Suzuki, Masahiko*; Blaber, M.; Kuroki, Ryota
Protein Science, 24(3), p.395 - 407, 2015/03
Times Cited Count:72 Percentile:89.80(Biochemistry & Molecular Biology)UDP-glucose: anthocyanidin 3--glucosyltransferase (UGT78K6) from catalyzes the transfer of glucose from UDP-glucose to anthocyanidins such as delphinidin. To understand the acceptor-recognition scheme of UGT78K6, the crystal structure of UGT78K6 and its complex forms with anthocyanidin delphinidin and petunidin, and flavonol kaempferol were determined to resolutions of 1.85  , 2.55 , 2.70 and 1.75 respectively. The anthocyanidin- and flavonol-acceptor binding details are almost identical in each complex structure, although the glucosylation activities against each acceptor were significantly different. The acceptor substrates in UGT78K6 are reversely bound to its binding site by a 180
, 2.55 , 2.70 and 1.75 respectively. The anthocyanidin- and flavonol-acceptor binding details are almost identical in each complex structure, although the glucosylation activities against each acceptor were significantly different. The acceptor substrates in UGT78K6 are reversely bound to its binding site by a 180 rotation about the O1-O3 axis of the flavonoid backbones observed in
rotation about the O1-O3 axis of the flavonoid backbones observed in 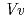 GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
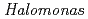 sp.593
sp.593Arai, Shigeki; Yonezawa, Yasushi*; Ishibashi, Matsujiro*; Matsumoto, Fumiko*; Adachi, Motoyasu; Tamada, Taro; Tokunaga, Hiroko*; Blaber, M.; Tokunaga, Masao*; Kuroki, Ryota
Acta Crystallographica Section D, 70(3), p.811 - 820, 2014/03
Times Cited Count:13 Percentile:63.41(Biochemical Research Methods)In order to clarify the structural basis of halophilic characteristics of an alkaline phosphatase derived from the moderate halophile sp.593 (HaAP), the tertiary structure of HaAP was determined to 2.1 resolution by X-ray crystallography. Structural properties of surface negative charge and core hydrophobicity are shown to be intermediate between halophile and non-halophile characteristics, and may explain the unique functional adaptation to a wide-range of salt concentration.
Adachi, Motoyasu; Shimizu, Rumi; Kuroki, Ryota; Blaber, M.
Journal of Synchrotron Radiation, 20(6), p.953 - 957, 2013/11
Times Cited Count:2 Percentile:12.49(Instruments & Instrumentation)Symfoil-4P is a 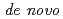 protein exhibiting the threefold symmetrical beta-trefoil fold designed based on the human acidic fibroblast growth factor. First three asparagine-glycine sequences of Symfoil-4P are replaced with glutamine-glycine (Symfoil-QG) or serine-glycine (Symfoil-SG) sequences protecting from deamidation, and His-Symfoil-II was prepared by introducing a protease digestion site into Symfoil-QG so that Symfoil-II has three complete repeats after removal of the N-terminal histidine tag. The Symfoil-QG and SG and His-Symfoil-II proteins were expressed in
protein exhibiting the threefold symmetrical beta-trefoil fold designed based on the human acidic fibroblast growth factor. First three asparagine-glycine sequences of Symfoil-4P are replaced with glutamine-glycine (Symfoil-QG) or serine-glycine (Symfoil-SG) sequences protecting from deamidation, and His-Symfoil-II was prepared by introducing a protease digestion site into Symfoil-QG so that Symfoil-II has three complete repeats after removal of the N-terminal histidine tag. The Symfoil-QG and SG and His-Symfoil-II proteins were expressed in 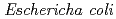 as soluble protein, and purified by nickel affinity chromatography. Symfoil-II was further purified by anion-exchange chromatography after removing the HisTag by proteolysis. Symfoil-QG and II crystals gave 1.5 and 1.1, resolution, respectively. The refined crystal structure of Symfoil-II showed pseudo-threefold symmetry as expected from other Symfoils.
as soluble protein, and purified by nickel affinity chromatography. Symfoil-II was further purified by anion-exchange chromatography after removing the HisTag by proteolysis. Symfoil-QG and II crystals gave 1.5 and 1.1, resolution, respectively. The refined crystal structure of Symfoil-II showed pseudo-threefold symmetry as expected from other Symfoils.
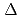 1-tetrahydrocannabinolic acid (THCA) synthase, the enzyme controlling the psychoactivity of
1-tetrahydrocannabinolic acid (THCA) synthase, the enzyme controlling the psychoactivity of 
Shoyama, Yoshinari*; Tamada, Taro; Kurihara, Kazuo; Takeuchi, Ayako*; Taura, Futoshi*; Arai, Shigeki; Blaber, M.*; Shoyama, Yukihiro*; Morimoto, Satoshi*; Kuroki, Ryota
Journal of Molecular Biology, 423(1), p.96 - 105, 2012/10
Times Cited Count:90 Percentile:89.94(Biochemistry & Molecular Biology)1-tetrahydrocannabinolic acid (THCA) synthase catalyzes the oxidative cyclization of cannabigerolic acid (CBGA) into THCA, the precursor of the primary psychoactive agent 1-tetrahydrocannabinol in . The structure-function relationship of THCA synthase was investigated by X-ray structure determination (2.75 resolution) and mutational analysis. Specific amino acid residues were identified in the active site of THCA synthase that are involved in the oxidative cyclization of the CBGA substrate.
sp. nucleoside diphosphate kinaseArai, Shigeki; Yonezawa, Yasushi; Okazaki, Nobuo; Matsumoto, Fumiko; Tamada, Taro; Tokunaga, Hiroko*; Ishibashi, Matsujiro*; Blaber, M.; Tokunaga, Masao*; Kuroki, Ryota
Protein Science, 21(4), p.498 - 510, 2012/04
Times Cited Count:15 Percentile:34.81(Biochemistry & Molecular Biology)In order to clarify the oligomer state of nucleoside diphosphate kinase (NDK) from moderately halophilic sp. 593 (HaNDK), the crystal structure of HaNDK was determined by X-ray crystallography. The crystal structures of the wild-type HaNDK and the mutant HaNDK (E134A) showed a dimer and a tetramer, respectively. The higher ordered association of proteins usually contributes to an increase in thermal stability and substrate affinity. The change in the assembly form by a minimum mutation may be an effective way for NDK to acquire molecular characteristics suited to various circumstances.
 KM1
KM1Okazaki, Nobuo; Tamada, Taro; Feese, M. D.*; Kato, Masaru*; Miura, Yutaka*; Komeda, Toshihiro*; Kobayashi, Kazuo*; Kondo, Keiji*; Blaber, M.*; Kuroki, Ryota
Protein Science, 21(4), p.539 - 552, 2012/04
Times Cited Count:5 Percentile:10.88(Biochemistry & Molecular Biology)Honjo, Eijiro; Tamada, Taro; Adachi, Motoyasu; Kuroki, Ryota; Meher, A.*; Blaber, M.*
Journal of Synchrotron Radiation, 15(3), p.285 - 287, 2008/05
Times Cited Count:5 Percentile:29.06(Instruments & Instrumentation)We attempt to improve a crystal contact of human acidic fibroblast growth factor (haFGF1) to control the crystal growth because haFGF crystallizes only as a thin-plate form. X-ray crystal analysis of haFGF showed that side chain Glu81, located at a crystal contact between haFGF molecules related by crystallographic symmetry, were in close proximity, suggesting that charge-repulsion may disrupt suitable crystal-packing interaction. To investigate whether the Glu residue affects crystal packing, we constructed haFGF mutants. Although crystals of Ala, Val and Leu mutants were grown as a thin-plate form by the same precipitant (formate) as wild type, crystals of Ser and Thr mutants were grown as a more bulky form. X-ray structural analysis of Ser and Thr mutants determined at 1.5 resolution revealed that hydroxyl groups of Ser and Thr were linked by hydrogen bonds mediated by formate used as a precipitant.
Okazaki, Nobuo; Ohara, Takashi; Umino, Hisao*; Chatake, Toshiyuki*; Kurihara, Kazuo; Cachau, R. E.*; Blaber, M.*; Niimura, Nobuo*; Kuroki, Ryota
no journal, ,
Shimizu, Rumi; Adachi, Motoyasu; Kuroki, Ryota; Blaber, M.
no journal, ,
We have already succeeded in creation of the de novo designed protein (Symfoil) exhibiting the threefold symmetrical -trefoil fold based on the human acidic fibroblast growth factor. Based on the Symfoil protein, we created Symfoil-II having the three repeats. The Symfoil-II protein was expressed in Eschericha coli as soluble protein, and purified by metal affinity chromatography using nickel-chelated agarose. The Symfoil-II was further purified by anion-exchange column chromatography after removing the HisTag by proteolysis. Symfoil-II was crystallized in 0.1 M Tris-HCl buffer (pH7.0) containing 1.8 M ammonium sulfate as precipitant at 20C. The X-ray diffraction data was collected to 1.9 resolution using a Rigaku R-AXIS VII diffractometer. The crystal belongs to a monoclinic space group C2. The refined crystal structure of Symfoil-II showed three fold symmetry as is observed in other Symfoils.
Adachi, Motoyasu; Okazaki, Nobuo*; Tamada, Taro; Kato, Masaru*; Kobayashi, Kazuo*; Blaber, M.; Kuroki, Ryota
no journal, ,
Maltooligosyl trehalose synthase (MTSase) and maltooligosyl trehalose trehalohydrolase (MTHase) are key enzymes for effective production of trehalose which is a useful compound as a preservative for foods. To understand the catalytic mechanism of MTSase, the crystal structure of MTSase derived from the hyperthermophilic archaeum Sulfolobus shibatae DSM5389 has been determined to 2.3 resolution by X-ray crystallography. Three invariant catalytic carboxylic amino acids in the  -amylase family are found in MTSase at positions Asp241, Glu269 and Asp460 in the (/)8 domain.
-amylase family are found in MTSase at positions Asp241, Glu269 and Asp460 in the (/)8 domain.
Adachi, Motoyasu; Honjo, Eijiro; Tamada, Taro; Blaber, M.*; Kuroki, Ryota
no journal, ,
Human acidic fibroblast growth factor (haFGF) is a powerful mitogen and angiogenic factor. Our objective is to reveal structure function relationships of haFGF by protein neutron crystallography. For the determination of neutron crystal structures, large crystal in size is required. Since haFGF forms thin crystals, we tried improvement of crystal growth by changing amino acids at molecular interface. The analysis using X-ray crystal structure showed that the residue of Glu81 is located near Glu81 generated by crystallographic symmetry. Therefore, Glu81 was replaced with Ala, Val, Leu, Ser and Thr residue. The mutants of E81S and E81T provided larger crystal in size than that of WT. The X-ray structures of E81S and E81T showed that a molecule of formic acid bridges between the two Glu81 residues by hydrogen bonding.
Okazaki, Nobuo; Ohara, Takashi; Umino, Hisao*; Chatake, Toshiyuki*; Kurihara, Kazuo; Cachau, R. E.*; Blaber, M.*; Niimura, Nobuo*; Kuroki, Ryota
no journal, ,
In protein molecules, key energetic contributors are solvation, desolvation and hydrogen bonding. They contribute protein folding, dynamics and molecular recognition. As a result, more elaborate studies of hydrogen atoms will be great help to recognize protein structures and obtain new findings of them. However, we do not have system which dedicated to characterization and analysis of hydrogen bonding. Therefore, we have developed a database for hydrogen and hydration water molecules. That database named Hydrogen and Hydration Database for Biomolecules (HHDB). Hydrogen bond data stored to HHDB use hydrogen atom coordinates determined directly by neutron diffraction and certain extremely high resolution X-ray diffraction. HHDB provides graphical user interface, users can use it through web browser. HHDB can visualize hydrogen atom positions in protein and solvent, and hydrogen bonding interactions. Fig. 1 shows HHDB plot example. In this plot, hydrogen atom is placed at the origin, and each point represents hydrogen bond distance and angle. We are improving the web user interfaces and the performance for usability.
Adachi, Motoyasu; Shimizu, Rumi; Kuroki, Ryota; Blaber, M.
no journal, ,
We have already succeeded in creation of the de novo designed protein (Symfoil) exhibiting the threefold symmetrical -trefoil fold based on the human acidic fibroblast growth factor. Based on the Symfoil protein, we created Symfoil-II having the three repeats. The Symfoil-II protein was expressed in Eschericha coli as soluble protein, and purified by metal affinity chromatography using nickel-chelated agarose. The Symfoil-II was further purified by anion-exchange column chromatography after removing the HisTag by proteolysis. Symfoil-II was crystallized in 0.1 M Tris-HCl buffer (pH7.0) containing 1.8 M ammonium sulfate as precipitant at 20C. The X-ray diffraction data was collected to 1.9 resolution using a Rigaku R-AXIS VII diffractometer. The crystal belongs to a monoclinic space group C2. The refined crystal structure of Symfoil-II showed three fold symmetry as is observed in other Symfoils.
