


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Ikeda, Takashi; Boero, M.*
Journal of Chemical Physics, 143(19), p.194510_1 - 194510_7, 2015/11
Times Cited Count:32 Percentile:76.46(Chemistry, Physical)By resorting to a novel implementation of the first-principles-based van der Waals correction based on maximally localized Wannier functions, we inspect its performance and assess its reliability for aqueous solutions of alkali metal ions. We find that van der Waals interactions, when added to the widely used revPBE gradient corrected functional, influence substantially both structural and dynamical properties of water molecules, with particular emphasis on the hydration shell of the alkali cations. These effects are more evident for strong structure-making and -breaking cationic species. Moreover, self-diffusion coefficients and reorientation correlation times of solvating water molecules change systematically, showing a trend in better agreement with experiments with respect to simulations neglecting the long-range dispersion contributions.
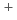 and Cs
and CsIkeda, Takashi; Boero, M.*
Journal of Chemical Physics, 137(4), p.041101_1 - 041101_4, 2012/07
Times Cited Count:30 Percentile:72.16(Chemistry, Physical)Hydration structure and polarization of Rb and Cs in liquid water at ambient conditions were studied by first principles molecular dynamics. Our systematic analysis of the relevant electronic structures, based on maximally localized Wannier functions, revealed that the dipole moment of H
and Cs in liquid water at ambient conditions were studied by first principles molecular dynamics. Our systematic analysis of the relevant electronic structures, based on maximally localized Wannier functions, revealed that the dipole moment of H O molecules in the first solvation shell of the ions slightly increases with increasing the atomic number. We also found that the polarization of heavy alkali ions, particularly Cs, tends to stabilize a peculiar asymmetric hydration structure with relevant consequences in the extraction of the harmful
O molecules in the first solvation shell of the ions slightly increases with increasing the atomic number. We also found that the polarization of heavy alkali ions, particularly Cs, tends to stabilize a peculiar asymmetric hydration structure with relevant consequences in the extraction of the harmful 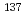 Cs resulting from nuclear wastes.
Cs resulting from nuclear wastes.
Wang, X.*; Hou, Z.*; Ikeda, Takashi; Huang, S.-F.*; Terakura, Kiyoyuki*; Boero, M.*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
Physical Review B, 84(24), p.245434_1 - 245434_7, 2011/12
Times Cited Count:33 Percentile:74.81(Materials Science, Multidisciplinary)The structural and electronic properties of N-doped zigzag graphene ribbons with various ratios of di- to monohydrogenated edge carbons are investigated within the density functional theory framework. We find that the stability of graphitic N next to the edge, which is claimed to play important roles in the catalytic activity in our previous work, will be enhanced with increasing the concentration of di-hydrogenated carbons. Furthermore, the di-hydrogenated edge carbons turn out to be easily converted into mono-hydrogenated ones in the presence of oxygen molecules at room temperature. Based on our results, we propose a possible way to enhance the oxygen reduction catalytic activity of N-doped graphene by controlling the degrees of hydrogenation of edge carbons. The characteristic features in the X-ray absorption and emission spectra for each specific N site considered here will also be given.
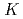 -edge X-ray absorption spectra of nanographene
-edge X-ray absorption spectra of nanographeneHou, Z.*; Wang, X.*; Ikeda, Takashi; Huang, S.-F.*; Terakura, Kiyoyuki*; Boero, M.*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
Journal of Physical Chemistry C, 115(13), p.5392 - 5403, 2011/03
Times Cited Count:38 Percentile:70.69(Chemistry, Physical)Carbon 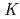 -edge X-ray absorption spectra of nanographene have been simulated by density functional theory calculations in order to obtain the information on the edge termination by hydrogen. Our results show that different edge terminations significantly affect the binding energy of 1s core-level of C atoms in the vicinity of edges because of the change in chemical bonding and the localized edge states. We find that a shoulder or a peak appears below the
-edge X-ray absorption spectra of nanographene have been simulated by density functional theory calculations in order to obtain the information on the edge termination by hydrogen. Our results show that different edge terminations significantly affect the binding energy of 1s core-level of C atoms in the vicinity of edges because of the change in chemical bonding and the localized edge states. We find that a shoulder or a peak appears below the 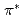 peak at relatively different positions with respect to the peak position in the theoretical spectra of zigzag graphene nano-ribbons, depending on the ratio of mono-hydrogen- to di-hydrogen-terminations. We also point out that the two additional features observed between the and
peak at relatively different positions with respect to the peak position in the theoretical spectra of zigzag graphene nano-ribbons, depending on the ratio of mono-hydrogen- to di-hydrogen-terminations. We also point out that the two additional features observed between the and 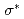 peaks of an ideal graphene originate from the states of C-H bonding and C-H
peaks of an ideal graphene originate from the states of C-H bonding and C-H bonding at the edges.
bonding at the edges.
Ikeda, Takashi; Boero, M.*; Huang, S.-F.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Ozaki, Junichi*; Miyata, Seizo*
Journal of Physical Chemistry C, 114(19), p.8933 - 8937, 2010/05
Times Cited Count:64 Percentile:82.84(Chemistry, Physical)Carbon Alloy Catalysts (CACs) have been attracting a growing interest as potential Pt-free electrode catalysts for polymer electrolyte fuel cell. In this computational study, we inspect possible oxygen adsorption and reduction processes on various models for exposed edges of these catalysts via first principles molecular dynamics. Our simulations suggest that the codoping of boron and nitrogen in CACs is a promising route to further enhancement of their catalytic activity with respect to both stability and reactivity.
Ikeda, Takashi; Huang, S.-F.*; Boero, M.*; Terakura, Kiyoyuki*
Hakkin Daitai Kabon Aroi Shokubai, p.121 - 138, 2010/04
no abstracts in English
Huang, S.-F.*; Terakura, Kiyoyuki*; Ozaki, Taisuke*; Ikeda, Takashi; Boero, M.*; Oshima, Masaharu*; Ozaki, Junichi*; Miyata, Seizo*
Physical Review B, 80(23), p.235410_1 - 235410_12, 2009/12
Times Cited Count:174 Percentile:97.23(Materials Science, Multidisciplinary)Recent studies suggest that the carbon-alloy catalyst with doped nitrogen may be a powerful candidate for cathode catalyst of fuel cell. In this paper, we aim to clarify the microscopic mechanisms of the enhancement in the catalyst activity caused by nitrogen doping using a simple graphene cluster model. We analyze modifications in the electronic structures and the energetical stability for some different configurations of N doping. We extend the analysis to the case of co-doping of nitrogen and boron and propose two possible scenarios explaining the further enhancement of catalytic activity by N and B co-doping.
Ikeda, Takashi; Huang, S.-F.*; Boero, M.*; Terakura, Kiyoyuki*
Gurafuen No Kino To Oyo Tembo, p.46 - 59, 2009/07
no abstracts in English
Ikeda, Takashi; Boero, M.*; Morikawa, Yoshitada*
Nihon Butsuri Gakkai-Shi, 64(4), p.256 - 262, 2009/04
First-principles-based simulations not relying on any empirical parameters are nowadays applicable to elucidate elemental processes of various chemical reactions occurring in condensed phase as well as in gas phase. In this article, the present status of the reactive simulations for surface, interface and biological systems is reviewed as examples and their future subjects are also summarized.
Ikeda, Takashi; Boero, M.*; Huang, S.-F.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Ozaki, Junichi*
Journal of Physical Chemistry C, 112(38), p.14706 - 14709, 2008/09
Times Cited Count:460 Percentile:99.36(Chemistry, Physical)Nitrogen-doped carbon-based catalysts are attracting a renovated interest as potential Pt-free electrode catalysts for polymer electrolyte fuel cell. In this computational study, we inspect possible oxygen adsorption and reduction processes on various models for the exposed edges of these catalysts. The dynamics of an O molecule solvated in water, mimicking the cathode environment, shows that O adsorption depends on the morphology and atomic structure of the system. We show that carbon alloys with N dopants at specific sites can exhibit a metal-free catalytic activity.
Ikeda, Takashi; Boero, M.*; Terakura, Kiyoyuki*
Journal of Chemical Physics, 127(7), p.074503_1 - 074503_8, 2007/08
Times Cited Count:153 Percentile:97.67(Chemistry, Physical)We studied the solvation structures of the divalent metal cations Mg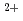 and Ca in ambient water by applying a Car-Parrinello-based constrained molecular dynamics method. By employing the metal-water oxygen coordination number as a reaction coordinate, we could identify distinct aqua complexes characterized by structural variations of the first coordination shell. In particular, our estimated free-energy profile clearly shows that the global minimum for Mg is represented by a rather stable six-fold coordination in the octahedral arrangement, in agreement with experiments. Conversely, for Ca the free-energy curve shows several shallow local minima, suggesting that the hydration structure of Ca is highly variable. Implications for water exchange reactions are also discussed.
and Ca in ambient water by applying a Car-Parrinello-based constrained molecular dynamics method. By employing the metal-water oxygen coordination number as a reaction coordinate, we could identify distinct aqua complexes characterized by structural variations of the first coordination shell. In particular, our estimated free-energy profile clearly shows that the global minimum for Mg is represented by a rather stable six-fold coordination in the octahedral arrangement, in agreement with experiments. Conversely, for Ca the free-energy curve shows several shallow local minima, suggesting that the hydration structure of Ca is highly variable. Implications for water exchange reactions are also discussed.
Boero, M.*; Ikeda, Takashi; Hirata, Masaru
Annual Report of the Earth Simulator Center April 2005 - March 2006, p.267 - 270, 2007/01
no abstracts in English
Ikeda, Takashi; Boero, M.*; Terakura, Kiyoyuki*
Journal of Chemical Physics, 126(3), p.034501_1 - 034501_9, 2007/01
Times Cited Count:148 Percentile:97.59(Chemistry, Physical)Structural and dynamical properties of the hydration of Li, Na, and K in liquid water at ambient conditions were studied by first principles molecular dynamics. Our simulations successfully captured the different hydration behavior shown by the three alkali ions as observed in experiments. The present analyses of the dependence of the self-diffusion coefficient and rotational correlation time of water on the ion concentration suggest that Li (K) is certainly categorized to structure maker (breaker), whereas Na acts as a weak structure breaker. An analysis of the relevant electronic structures, based on maximally localized Wannier functions, revealed that the dipole moment of HO molecules in the first solvation shell of Na and Kdecreases by about 0.1 Debye compared to that in the bulk, due to a contraction of the oxygen lone pair orbital pointing toward the metal ion.
and Mg metal cations in the hydrolysis reactionBoero, M.*; Ikeda, Takashi; Ito, Etsuro*; Terakura, Kiyoyuki*
Journal of the American Chemical Society, 128(51), p.16798 - 16807, 2006/12
Times Cited Count:55 Percentile:76.63(Chemistry, Multidisciplinary)Hybrid quantum-mechanics/molecular mechanics simulations, coupled to the recently introduced metadynamics method, performed on the adenosine triphosphate (ATP) of the bovine Hsc70 ATPase protein, show which specific water molecule of the solvation shell of the Mg metal cation acts as a trigger in the initial phase of the ATP hydrolysis reaction in ATP synthase. Furthermore, we provide a detailed picture of the reaction mechanism, not accessible to experimental probes.
Ikeda, Takashi; Boero, M.*; Terakura, Kiyoyuki*
no journal, ,
We studied the solvation structures of the divalent metal cations Mg and Ca in ambient water by applying a Car-Parrinello-based constrained molecular dynamics method. By employing the metal-water oxygen coordination number as a reaction coordinate, we could identify distinct aqua complexes characterized by structural variations of the first coordination shell. In particular, our estimated free-energy profile clearly shows that the global minimum for Mg is represented by a rather stable six-fold coordination in the octahedral arrangement, in agreement with experiments. Conversely, for Ca the free-energy curve shows several shallow local minima, suggesting that the hydration structure of Ca is highly variable. Implications for water exchange reactions are also discussed.
Ikeda, Takashi; Boero, M.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Ozaki, Junichi*
no journal, ,
no abstracts in English
Ikeda, Takashi; Boero, M.*; Huang, S.-F.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Ozaki, Junichi*; Miyata, Seizo*
no journal, ,
no abstracts in English
