


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Chiba, Kaori*; Matsui, Takuro*; Chatake, Toshiyuki*; Ohara, Takashi; Tanaka, Ichiro*; Yutani, Katsuhide*; Niimura, Nobuo*
Protein Science, 32(10), p.e4765_1 - e4765_13, 2023/10
Times Cited Count:0 Percentile:0(Biochemistry & Molecular Biology)Chatake, Toshiyuki*; Fujiwara, Satoru
Acta Crystallographica Section D; Structural Biology (Internet), 72(1), p.71 - 82, 2016/01
Times Cited Count:5 Percentile:37.86(Biochemical Research Methods) and CaCl
and CaClChatake, Toshiyuki*; Sunami, Tomoko
Journal of Inorganic Biochemistry, 124, p.15 - 25, 2013/07
Times Cited Count:16 Percentile:60.13(Biochemistry & Molecular Biology)Chatake, Toshiyuki*; Sazaki, Gen*; Kikko, Tatsuhiko*; Fujiwara, Satoru; Ishikawa, Takuya*; Matsumoto, Osamu*; Morimoto, Yukio*
Crystal Growth & Design, 10(3), p.1090 - 1095, 2010/03
Times Cited Count:4 Percentile:48.31(Chemistry, Multidisciplinary)We propose a technique for DNA crystallization using the thermal reversible process of DNA: a conversion between a double-stranded DNA (dsDNA) and two single-stranded DNAs (ssDNAs) with temperatures. We investigated the solubility of the crystals of a DNA hexamer d(CGCGCG) and their melting temperature, at which the thermal conversion occurs. The results obtained suggest that the conversion from a dsDNA to ssDNAs results in an increase in solubility. It was shown that using this temperature-controlled technique, high-grade single crystals of the DNA hexamer could be obtained from a small amount of DNA samples. This easy-to-apply technique would be superior to the conventional vapor diffusion technique in that it allows the solubility of DNA crystals to be controlled with no need for expensive setups.
Matsumoto, Fumiko; Maeda, Kayo*; Chatake, Toshiyuki*; Maeda, Yuichiro*; Fujiwara, Satoru
Biochemical and Biophysical Research Communications, 382(1), p.205 - 209, 2009/04
Times Cited Count:15 Percentile:37.83(Biochemistry & Molecular Biology)Two cardiomyopathy-causing mutations, E244D and K247R, in human cardiac troponin T (TnT) are located in the coiled-coil region of the Tn-core domain. To elucidate effects of mutations in this region on the regulatory function of Tn, we measured Ca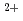 -dependent ATPase activity of myofibrils containing various mutants of TnT at these residues. The results confirmed that the mutant E244D increases the maximum ATPase activity without changing the Ca sensitivity. The mutant K247R was shown for the first time to have the effect similar to the mutant E244D. Furthermore, various TnT mutants (E244D, E244M, E244A, E244K, K247R, K247E, and K247A) showed various effects on the maximum ATPase activity while the Ca sensitivity was unchanged. Molecular dynamics simulations of the Tn-core containing these TnT mutants suggested that the hydrogen-bond network formed by the side chains of neighboring residues around residues 244 and 247 is important for Tn to function properly.
-dependent ATPase activity of myofibrils containing various mutants of TnT at these residues. The results confirmed that the mutant E244D increases the maximum ATPase activity without changing the Ca sensitivity. The mutant K247R was shown for the first time to have the effect similar to the mutant E244D. Furthermore, various TnT mutants (E244D, E244M, E244A, E244K, K247R, K247E, and K247A) showed various effects on the maximum ATPase activity while the Ca sensitivity was unchanged. Molecular dynamics simulations of the Tn-core containing these TnT mutants suggested that the hydrogen-bond network formed by the side chains of neighboring residues around residues 244 and 247 is important for Tn to function properly.
Ishikawa, Takuya*; Chatake, Toshiyuki*; Onishi, Yuki*; Tanaka, Ichiro*; Kurihara, Kazuo; Kuroki, Ryota; Niimura, Nobuo*
Chemical Physics, 345(2-3), p.152 - 158, 2008/04
Times Cited Count:14 Percentile:44.65(Chemistry, Physical)Chatake, Toshiyuki*; Shibayama, Naoya*; Park, S. Y.*; Kurihara, Kazuo; Tamada, Taro; Tanaka, Ichiro*; Niimura, Nobuo*; Kuroki, Ryota; Morimoto, Yukio*
Journal of the American Chemical Society, 129(48), p.14840 - 14841, 2007/12
Times Cited Count:25 Percentile:60.26(Chemistry, Multidisciplinary)Arai, Shigeki; Chatake, Toshiyuki*; Niimura, Nobuo
Nihon Kessho Gakkai-Shi, 48(2), p.133 - 139, 2006/04
It has long been suspected that the structure and function of a DNA duplex can be strongly dependent on its degree of hydration. By neutron diffraction experiments, we have succeeded in determining most of the hydrogen (H) and deuterium (D) atomic positions in the d(CCATTAATGG) duplex. Moreover, the D positions in 27 DO molecules have been determined. In particular, the complex water network in the minor groove has been observed in detail. By a combined structural analysis using 2.0 AA resolution X-ray and 3.0 AA resolution neutron data, it is clear that the spine of hydration is built up, not only by a simple hexagonal hydration pattern (as reported in prior X-ray studies), but also by many other water bridges hydrogen-bonded to the DNA strands. The complexity of the hydration pattern in the minor groove is derived from an extraordinary variety of orientations displayed by the water molecules.
Niimura, Nobuo; Arai, Shigeki; Kurihara, Kazuo; Chatake, Toshiyuki*; Tanaka, Ichiro*; Bau, R.*
Cellular and Molecular Life Sciences, 63(3), p.285 - 300, 2006/02
Times Cited Count:41 Percentile:37.86(Biochemistry & Molecular Biology)Neutron diffraction provides an experimental method of directly locating hydrogen atoms in proteins and DNA oligomers. Three different types of high resolution neutron diffractometers for biological macromolecules have been constructed in Japan, France and the U.S.A., and they have all been actively used in recent years to determine the crystal structures of numerous proteins. Examples include the detailed geometries of hydrogen bonds, information on H/D exchange in proteins, the unambiguous location of protons, the role of key hydrogen atoms in enzymatic activity and thermostability, and the dynamical behavior of hydration structures, all of which have been extracted from these structural results and reviewed in this article. Other important techniques, such as the optimization of growth of large single crystals using phase diagrams, the preparation of fully deuterated proteins, the introduction of cryogenic techniques to neutron protein crystallography, and the establishment of a "hydrogen and hydration in proteins" database, will also be described in this paper.
observed by neutron diffraction measurementsArai, Shigeki; Chatake, Toshiyuki*; Ohara, Takashi; Kurihara, Kazuo; Tanaka, Ichiro*; Suzuki, Nobuhiro*; Fujimoto, Zui*; Mizuno, Hiroshi*; Niimura, Nobuo
Nucleic Acids Research, 33(9), p.3017 - 3024, 2005/05
Times Cited Count:93 Percentile:82.96(Biochemistry & Molecular Biology)It has long been suspected that the structure and function of a DNA duplex can be strongly dependent on its degree of hydration. By neutron diffraction experiments, we have succeeded in determining most of the hydrogen (H) and deuterium (D) atomic positions in the d(CCATTAATGG) duplex. Moreover, the D positions in 27 DO molecules have been determined. In particular, the complex water network in the minor groove has been observed in detail. By a combined structural analysis using 2.0 Å resolution X-ray and 3.0 Å resolution neutron data, it is clear that the spine of hydration is built up, not only by a simple hexagonal hydration pattern (as reported in prior X-ray studies), but also by many other water bridges hydrogen-bonded to the DNA strands. The complexity of the hydration pattern in the minor groove is derived from an extraordinary variety of orientations displayed by the water molecules.
Niimura, Nobuo; Arai, Shigeki; Kurihara, Kazuo; Chatake, Toshiyuki*; Tanaka, Ichiro*; Bau, R.*
Hydrogen- and Hydration-Sensitive Structural Biology, p.17 - 35, 2005/00
At the JAERI, we have constructed several high-resolution neutron diffractometers dedicated to biological macromolecules (called BIX-type diffractometers), which use a monochromatized neutron beam and a neutron imaging plate detector. In this paper, we review several interesting results regarding hydrogen positions and hydration in proteins, obtained using the two BIX-type diffractometers in JAERI. The general subject of neutron protein crystallography has been reviewed by several authors, and several selected topics have been discussed.
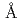 resolution
resolutionChatake, Toshiyuki*; Kurihara, Kazuo; Tanaka, Ichiro*; Tsyba, I.*; Bau, R.*; Jenney, F. E. Jr.*; Adams, M. W. W.*; Niimura, Nobuo
Acta Crystallographica Section D, 60(8), p.1364 - 1373, 2004/08
Times Cited Count:34 Percentile:88.89(Biochemical Research Methods)A neutron diffraction study has been carried out at 1.6 resolution on a mutant rubredoxin from 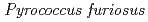 using the BIX-3 single-crystal diffractometer at the JRR-3 reactor of JAERI. In order to study the unusual thermostability of rubredoxin from
using the BIX-3 single-crystal diffractometer at the JRR-3 reactor of JAERI. In order to study the unusual thermostability of rubredoxin from 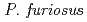 , the hydrogen-bonding patterns were compared between the native and a 'triple-mutant' variant where three residues were changed so that they are identical to those in a mesophilic rubredoxin. In the present study, some minor changes were found between the wild-type and mutant proteins in the hydrogen-bonding patterns of the Trp3/Tyr3 region. The H/D-exchange ratios in the protein were also studied. The results suggest that the backbone amide bonds near the four Cys residues of the FeS
, the hydrogen-bonding patterns were compared between the native and a 'triple-mutant' variant where three residues were changed so that they are identical to those in a mesophilic rubredoxin. In the present study, some minor changes were found between the wild-type and mutant proteins in the hydrogen-bonding patterns of the Trp3/Tyr3 region. The H/D-exchange ratios in the protein were also studied. The results suggest that the backbone amide bonds near the four Cys residues of the FeS redox center are most resistant to H/D exchange. In addition, the 1.6 resolution of the present neutron structure determination has revealed a more detailed picture than previously available of some portions of the water structure, including ordered and disordered O-D bonds.
redox center are most resistant to H/D exchange. In addition, the 1.6 resolution of the present neutron structure determination has revealed a more detailed picture than previously available of some portions of the water structure, including ordered and disordered O-D bonds.
by BIX-3, a single-crystal diffractometer for biomacromoleculesKurihara, Kazuo; Tanaka, Ichiro*; Chatake, Toshiyuki*; Adams, M. W. W.*; Jenney, F. E. Jr.*; Moiseeva, N.*; Bau, R.*; Niimura, Nobuo
Proceedings of the National Academy of Sciences of the United States of America, 101(31), p.11215 - 11220, 2004/08
Times Cited Count:48 Percentile:61.13(Multidisciplinary Sciences)The structure of a rubredoxin (Rd) from , an organism that grows optimally at 100  C, was determined using the neutron single-crystal diffractometer for biological macromolecules (BIX-3) at the JRR-3 reactor of JAERI. Data were collected at room temperature up to a resolution of 1.5 , and the completeness of the data set was 81.9 %. The model contains 306 H atoms and 50 D atoms. A total of 37 hydration water molecules were identified. The model has been refined to final agreement factors of
C, was determined using the neutron single-crystal diffractometer for biological macromolecules (BIX-3) at the JRR-3 reactor of JAERI. Data were collected at room temperature up to a resolution of 1.5 , and the completeness of the data set was 81.9 %. The model contains 306 H atoms and 50 D atoms. A total of 37 hydration water molecules were identified. The model has been refined to final agreement factors of 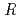 = 18.6 % and
= 18.6 % and 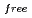 = 21.7 %. Several orientations of the O-D bonds of side chains, whose assignments from X-ray data were previously ambiguous, were clearly visible in the neutron structure. While most backbone N-H bonds had undergone some degree of H/D exchange throughout the molecule, five H atom positions still had distinctly negative (H) peaks. The neutron Fourier maps clearly showed the details of an extensive set of H bonds involving the ND
= 21.7 %. Several orientations of the O-D bonds of side chains, whose assignments from X-ray data were previously ambiguous, were clearly visible in the neutron structure. While most backbone N-H bonds had undergone some degree of H/D exchange throughout the molecule, five H atom positions still had distinctly negative (H) peaks. The neutron Fourier maps clearly showed the details of an extensive set of H bonds involving the ND
 terminus that may contribute to the unusual thermostability of this molecule.
terminus that may contribute to the unusual thermostability of this molecule.
Arai, Shigeki; Chatake, Toshiyuki; Suzuki, Nobuhiro*; Mizuno, Hiroshi*; Niimura, Nobuo
Acta Crystallographica Section D, 60(6), p.1032 - 1039, 2004/06
Times Cited Count:17 Percentile:76.04(Biochemical Research Methods)The parameters used for evaluating biomacromolecular crystal quality (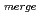 ,
, 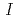 /
/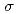 (), maximum resolution and mosaicity) strongly depend on the diffraction experimental conditions. In this paper we describe the distinctive features of the relative Wilson plot method, and we show that the overall B-factor obtained from this plot is given as a more appropriate to characterize protein crystals. The relative Wilson plot has been applied to the characterization of crystals of a B-DNA decamer d(CCATTAATGG), and crystals of the proteins DsrD (dissimilatory sulfite reductase D) and hen egg-white lysozyme (HEWL) which we have studied by neutron diffraction. We have found that the crystal qualities of the B-DNA decamer and DsrD significantly depend on the regions of the crystallization phase diagram from which samples were taken. However, in the case of HEWL, crystal quality appears to be independent on the region of the crystallization phase diagram.
(), maximum resolution and mosaicity) strongly depend on the diffraction experimental conditions. In this paper we describe the distinctive features of the relative Wilson plot method, and we show that the overall B-factor obtained from this plot is given as a more appropriate to characterize protein crystals. The relative Wilson plot has been applied to the characterization of crystals of a B-DNA decamer d(CCATTAATGG), and crystals of the proteins DsrD (dissimilatory sulfite reductase D) and hen egg-white lysozyme (HEWL) which we have studied by neutron diffraction. We have found that the crystal qualities of the B-DNA decamer and DsrD significantly depend on the regions of the crystallization phase diagram from which samples were taken. However, in the case of HEWL, crystal quality appears to be independent on the region of the crystallization phase diagram.
Niimura, Nobuo; Chatake, Toshiyuki; Kurihara, Kazuo; Maeda, Mitsuru
Cell Biochemistry and Biophysics, 40(3), p.351 - 369, 2004/06
Times Cited Count:24 Percentile:25.29(Biochemistry & Molecular Biology)Neutron diffraction provides an experimental method of directly locating hydrogen atoms in proteins. High resolution neutron diffractometers dedicated to biological macromolecules (BIX-type diffractometer) have been constructed at the Japan Atomic Energy Research Institute (JAERI) and they have been used in the 1.5 AA -resolution crystal structure analyses of several proteins.
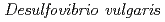
Chatake, Toshiyuki; Mizuno, Nobuhiro*; Voordown, G.*; Higuchi, Yoshiki*; Arai, Shigeki; Tanaka, Ichiro; Niimura, Nobuo
Acta Crystallographica Section D, 59(Part2), p.2306 - 2309, 2003/12
Times Cited Count:19 Percentile:75.55(Biochemical Research Methods)Issimilatory sulfite reductase D (DsrD) from has been crystallized for a neutron diffraction study. The initial crystals obtained were too small for the neutron experiment. In order to obtain a larger crystal (1 mm ), a combination of two techniques was used to find the optimum crystallization conditions: a crystallization phase diagram, followed by crystal-quality assessment via X-ray diffraction. Using conditions determined in this manner, a large single crystal (1.7 mm) of the DsrD protein was subsequently grown in DO solution by the macro-seeding technique. The neutron diffraction experiment was carried out using the BIX-3 diffractometer at the Japan Atomic Energy Research Institute (JAERI), and the diffraction data up to 2.4 AA resolution could be collected from this crystal.
), a combination of two techniques was used to find the optimum crystallization conditions: a crystallization phase diagram, followed by crystal-quality assessment via X-ray diffraction. Using conditions determined in this manner, a large single crystal (1.7 mm) of the DsrD protein was subsequently grown in DO solution by the macro-seeding technique. The neutron diffraction experiment was carried out using the BIX-3 diffractometer at the Japan Atomic Energy Research Institute (JAERI), and the diffraction data up to 2.4 AA resolution could be collected from this crystal.
Niimura, Nobuo; Chatake, Toshiyuki; Ostermann, A.; Kurihara, Kazuo; Tanaka, Ichiro
Zeitschrift f r Kristallographie, 218(2), p.96 - 107, 2003/03
r Kristallographie, 218(2), p.96 - 107, 2003/03
no abstracts in English
Chatake, Toshiyuki; Ostermann, A.; Kurihara, Kazuo; Parak, F.*; Niimura, Nobuo
Proteins: Structure, Function, and Bioinformatics, 50(3), p.516 - 523, 2003/02
Times Cited Count:54 Percentile:76.77(Biochemistry & Molecular Biology)no abstracts in English
Niimura, Nobuo; Chatake, Toshiyuki
Hamon, 13(1), p.47 - 50, 2003/01
no abstracts in English
Tanaka, Ichiro; Kurihara, Kazuo; Chatake, Toshiyuki; Niimura, Nobuo
Journal of Applied Crystallography, 35(Part1), p.34 - 40, 2002/02
A high performance neutron diffractometer for biological crystallography (BIX-3) has been constructed at JRR-3M in Japan Atomic Energy Research Institute (JAERI) in order to determine the hydrogen positions in biological macromolecules. It uses several recent technical innovations, such as a neutron imaging plate and an elastically bent silicon monochromator developed by the authors. These have made it possible to realize a compact vertical arrangement of the diffractometer. Diffraction data have been collected from the proteins rubredoxin and myoglobin in about one month, to a resolution of 1.5, good enough to identify the hydrogen atoms with high accuracy. By adopting a crystal-step scan method for measuring Bragg diffraction intensities, the signal to noise ratio was much better than that of the Laue method. This shows that BIX-3 is one of the best-performing machines for neutron protein crystallography in the world currently.
