


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 -glucosyltransferase from
-glucosyltransferase from 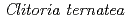
Hiromoto, Takeshi; Honjo, Eijiro*; Noda, Hisanobu*; Tamada, Taro; Kazuma, Kohei*; Suzuki, Masahiko*; Blaber, M.; Kuroki, Ryota
Protein Science, 24(3), p.395 - 407, 2015/03
Times Cited Count:72 Percentile:89.94(Biochemistry & Molecular Biology)UDP-glucose: anthocyanidin 3--glucosyltransferase (UGT78K6) from catalyzes the transfer of glucose from UDP-glucose to anthocyanidins such as delphinidin. To understand the acceptor-recognition scheme of UGT78K6, the crystal structure of UGT78K6 and its complex forms with anthocyanidin delphinidin and petunidin, and flavonol kaempferol were determined to resolutions of 1.85  , 2.55 , 2.70 and 1.75 respectively. The anthocyanidin- and flavonol-acceptor binding details are almost identical in each complex structure, although the glucosylation activities against each acceptor were significantly different. The acceptor substrates in UGT78K6 are reversely bound to its binding site by a 180
, 2.55 , 2.70 and 1.75 respectively. The anthocyanidin- and flavonol-acceptor binding details are almost identical in each complex structure, although the glucosylation activities against each acceptor were significantly different. The acceptor substrates in UGT78K6 are reversely bound to its binding site by a 180 rotation about the O1-O3 axis of the flavonoid backbones observed in
rotation about the O1-O3 axis of the flavonoid backbones observed in  GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
-glucosyltransferase from Hiromoto, Takeshi; Honjo, Eijiro*; Tamada, Taro; Noda, Hisanobu*; Kazuma, Kohei*; Suzuki, Masahiko*; Kuroki, Ryota
Journal of Synchrotron Radiation, 20(6), p.894 - 898, 2013/11
Times Cited Count:41 Percentile:86.70(Instruments & Instrumentation)Flowers of the butterfly pea () accumulate a group of polyacylated anthocyanins, named ternatins, in their petals. The first step in ternatin biosynthesis is the transfer of glucose from UDP-glucose to anthocyanidins such as delphinidin, a reaction catalyzed in  by UDP-glucose:anthocyanidin 3--glucosyltransferase (
by UDP-glucose:anthocyanidin 3--glucosyltransferase (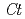 3GT-A; AB185904). To elucidate the structure-function relationship of 3GT-A, recombinant 3GT-A was expressed in
3GT-A; AB185904). To elucidate the structure-function relationship of 3GT-A, recombinant 3GT-A was expressed in 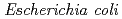 and its tertiary structure was determined to 1.85 , resolution by using X-ray crystallography. The structure of 3GT-A shows a common folding topology, the GT-B fold, comprised of two Rossmann-like
and its tertiary structure was determined to 1.85 , resolution by using X-ray crystallography. The structure of 3GT-A shows a common folding topology, the GT-B fold, comprised of two Rossmann-like 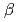 /
/ / domains and a cleft located between the N- and C-domains containing two cavities that are used as binding sites for the donor (UDP-Glc) and acceptor substrates. By comparing the structure of 3GT-A with that of the flavonoid glycosyltransferase GT1 from red grape (
/ domains and a cleft located between the N- and C-domains containing two cavities that are used as binding sites for the donor (UDP-Glc) and acceptor substrates. By comparing the structure of 3GT-A with that of the flavonoid glycosyltransferase GT1 from red grape (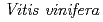 ) in complex with UDP-2-deoxy-2-fluoro glucose and kaempferol, locations of the catalytic His-Asp dyad and the residues involved in recognizing UDP-2-deoxy-2-fluoro glucose were essentially identical in 3GT-A, but certain residues of GT1 involved in binding kaempferol were found to be substituted in 3GT-A. These findings are important for understanding the differentiation of acceptor-substrate recognition in these two enzymes.
) in complex with UDP-2-deoxy-2-fluoro glucose and kaempferol, locations of the catalytic His-Asp dyad and the residues involved in recognizing UDP-2-deoxy-2-fluoro glucose were essentially identical in 3GT-A, but certain residues of GT1 involved in binding kaempferol were found to be substituted in 3GT-A. These findings are important for understanding the differentiation of acceptor-substrate recognition in these two enzymes.
Ito, Takachika*; Suzuki, Shoichi*; Kanaji, Sachiko*; Shiraishi, Hiroshi*; Ota, Shoichiro*; Arima, Kazuhiko*; Tanaka, Go*; Tamada, Taro; Honjo, Eijiro*; Garcia, K. C.*; et al.
Journal of Biological Chemistry, 284(36), p.24289 - 24296, 2009/09
Times Cited Count:24 Percentile:45.48(Biochemistry & Molecular Biology)Both IL-4 and IL-13 can bind to the shared receptor composed of the IL-4 receptor chain and the IL-13 receptor -1 chain (IL-13R1); however, the assembly mechanisms of these ligands to the receptor is different, enabling the principal functions of these ligands to be different. We have previously shown that the N-terminal Ig-like domain in IL-13R1, called the D1 domain, is the specific and critical binding unit for IL-13. However, it has still remained obscure which the amino acid has specific binding capacity to IL-13 and why the D1 domain acts as the binding site for IL-13, but not IL-4. To address these questions, in this study, we performed the mutational analyses for the D1 domain, combining the structural data to identify the amino acids critical for binding to IL-13. Mutations of Lys76, Lys77, or Ile78 in c' strand in which the crystal structure showed interact with IL-13 and those of Trp65 and Ala79 adjacent to the interacting site, resulted in significant impairment of IL-13 binding, demonstrating that these amino acids generate the binding site. Furthermore, mutations of Val35, Leu38, or Val42 at N-terminal -strand also resulted in loss of IL-13 binding, probably from decrease structural stability. None of the mutations employed here affected IL-4 binding. These results demonstrate that the hydrophobic patch composed of Lys76, Lys77, and Ile78 is the IL-13 recognition site and solidify our understanding that the differential requirements of the D1 domain in IL-13R1 allows the shared receptor to respond differentially to IL-4 and IL-13.
Adachi, Motoyasu; Ohara, Takashi; Kurihara, Kazuo; Tamada, Taro; Honjo, Eijiro; Okazaki, Nobuo; Arai, Shigeki; Shoyama, Yoshinari; Kimura, Kaname*; Matsumura, Hiroyoshi*; et al.
Proceedings of the National Academy of Sciences of the United States of America, 106(12), p.4641 - 4646, 2009/03
Times Cited Count:115 Percentile:90.49(Multidisciplinary Sciences)To further understand the catalytic mechanism and inhibitor recognition of HIV-1 protease, we need to determine the locations of key hydrogen atoms in the catalytic aspartates Asp25 and Asp125. The structure of HIV-1 protease in complex with transition-state analog KNI-272 was determined by combined neutron crystallography at 1.9 resolution and X-ray crystallography at 1.4 resolution. The resulting structural data shows that the catalytic residue Asp25 is protonated and that Asp125 is deprotonated. The proton on Asp25 makes a hydrogen bond with the carbonyl group of the allophenylnorstatine group in KNI-272. The deprotonated Asp125 bonds to the hydroxyl proton of Apns. The results provide direct experimental evidence for proposed aspects of the catalytic mechanism of HIV-1 protease; and can therefore contribute substantially to the development of specific inhibitors for therapeutic application.
chain and interleukin-13 receptor 1 chain by a silkworm-baculovirus systemHonjo, Eijiro; Shoyama, Yoshinari; Tamada, Taro; Shigematsu, Hideki*; Hatanaka, Takaaki*; Kanaji, Sachiko*; Arima, Kazuhiko*; Ito, Yuji*; Izuhara, Kenji*; Kuroki, Ryota
Protein Expression and Purification, 60(1), p.25 - 30, 2008/07
Times Cited Count:13 Percentile:32.58(Biochemical Research Methods)The receptor binding to Interleukin (IL)-13 is composed of the IL-13 receptor 1 chain (IL-13R 1) and the IL-4 receptor chain (IL-4R ). In order to investigate the interaction of IL-13 with IL-13R 1 and IL-4R , the DNA fragments coding the extracellular regions of human IL-13R 1 and the IL-4R were fused with mouse Fc and expressed by a silkworm-baculovirus system. The expressed receptors were successfully purified by affinity chromatography using protein A, and the Fc region was removed by thrombin digestion. Size exclusion chromatography and SPR analysis revealed that mixture of IL-13 and IL-13R1 showed predominant affinity to IL-4R, although neither detectable affinity of IL-13 nor IL-13R1 was observed against IL-4R. Combining these data with the moderate affinity of IL-13 to IL-13R1, this indicates that IL-13 first binds to IL-13R1 and recruits consequently to IL-4R.
Honjo, Eijiro; Tamada, Taro; Adachi, Motoyasu; Kuroki, Ryota; Meher, A.*; Blaber, M.*
Journal of Synchrotron Radiation, 15(3), p.285 - 287, 2008/05
Times Cited Count:5 Percentile:29.10(Instruments & Instrumentation)We attempt to improve a crystal contact of human acidic fibroblast growth factor (haFGF1) to control the crystal growth because haFGF crystallizes only as a thin-plate form. X-ray crystal analysis of haFGF showed that side chain Glu81, located at a crystal contact between haFGF molecules related by crystallographic symmetry, were in close proximity, suggesting that charge-repulsion may disrupt suitable crystal-packing interaction. To investigate whether the Glu residue affects crystal packing, we constructed haFGF mutants. Although crystals of Ala, Val and Leu mutants were grown as a thin-plate form by the same precipitant (formate) as wild type, crystals of Ser and Thr mutants were grown as a more bulky form. X-ray structural analysis of Ser and Thr mutants determined at 1.5 resolution revealed that hydroxyl groups of Ser and Thr were linked by hydrogen bonds mediated by formate used as a precipitant.
Honjo, Eijiro; Kuroki, Ryota; Kobori, Hiroshi*; Takakura, Hikaru*; Yahata, Kazuhide*; Sone, Takefumi*; Imamoto, Fumio*; Moriyama, Tatsuya*
Tampakushitsu Seisei To Toriatsukai No Kotsu, p.135 - 178, 2007/10
Protein expression and purification are important techniques for protein scientists. Especially in the field of molecular biology, the researchers often requires for some know-how on protein handling based on biochemical information. This book is aimed at introducing an overview of recombinant protein expression and purification, and furthermore providing experimental know-how about the selection of analytical reagents or instruments.
Yamada, Hidenori*; Tamada, Taro; Kosaka, Megumi*; Miyata, Kohei*; Fujiki, Shinya*; Tano, Masaru*; Moriya, Masayuki*; Yamanishi, Mamoru*; Honjo, Eijiro; Tada, Horiko*; et al.
Protein Science, 16(7), p.1389 - 1397, 2007/07
Times Cited Count:41 Percentile:60.18(Biochemistry & Molecular Biology)In an attempt to control protein incorporation in a crystal lattice, a leucine zipper-like hydrophobic interface (comprising four leucine residues) was introduced into a helical region (helix 2) of the human pancreatic ribonuclease 1 (RNase 1) that was predicted to form a suitable crystallization interface. Although crystallization of wild type RNase 1 has not yet been reported, the RNase 1 mutant having four leucines (4L-RNase 1) was successfully crystallized under several different conditions. The crystal structures were subsequently determined by X-ray crystallography by molecular replacement using the structure of bovine RNase A. The overall structure of 4L-RNase 1 is quite similar to that of the bovine RNase A, and the introduced leucine residues formed the designed crystal interface. To further characterize the role of the introduced leucine residues in crystallization of RNase 1, the number of leucines was reduced to three or two (3L- and 2L-RNase 1, respectively). Both mutants crystallized and a similar hydrophobic interface as in 4L-RNase 1 was observed. A related approach to engineer crystal contacts at helix 3 of RNase 1 (N4L-RNase 1) was also evaluated. N4L-RNase 1 also successfully crystallized, and formed the expected hydrophobic packing interface. These results suggest that appropriate introduction of a leucine zipper-like hydrophobic interface can promote intra molecular symmetry for more efficient protein crystallization in crystal lattice engineering efforts.
Tamada, Taro; Honjo, Eijiro; Kuroki, Ryota
Nihon Kessho Gakkai-Shi, 48(6), p.429 - 435, 2006/12
The crystal structure of the signaling complex of human granulocyte colony-stimulating factor (GCSF) and ligand binding region of human GCSF receptor (GCSF-R) has been determined to 2.8 resolution. The GCSF:GCSF-R complex formed a 2:2 stoichiometry via a cross-over interaction between the Ig-like domains of GCSF-R and GCSF. The conformation of the complex is quite different to that between human GCSF and the CRH domain of mouse GCSF-R. This cross-over homodimerization necessary for GCSF-R activation is consistent with previously reported thermodynamic and mutational studies.
Tamada, Taro; Honjo, Eijiro; Maeda, Yoshitake*; Okamoto, Tomoyuki*; Ishibashi, Matsujiro*; Tokunaga, Masao*; Kuroki, Ryota
Proceedings of the National Academy of Sciences of the United States of America, 103(9), p.3135 - 3140, 2006/02
Times Cited Count:99 Percentile:84.91(Multidisciplinary Sciences)A crystal structure of the signaling complex between human granulocyte colony-stimulating factor (GCSF), and a ligand binding region of GCSF receptor (GCSF-R), has been determined to 2.8 resolution. The GCSF:GCSF-R complex formed a 2:2 stoichiometry via a cross-over interaction between the Ig-like domains of GCSF-R and GCSF. The conformation of the complex is quite different to that between human GCSF and the CRH domain of mouse GCSF-R, but similar to that of the interleukin-6 (IL-6)/gp130 signaling complex. The Ig-like domain cross-over structure necessary for GCSF-R activation is consistent with previously reported thermodynamic and mutational analyses.
Honjo, Eijiro; Tamada, Taro; Maeda, Yoshitake*; Koshiba, Takumi*; Matsukura, Yasuko*; Okamoto, Tomoyuki*; Ishibashi, Matsujiro*; Tokunaga, Masao*; Kuroki, Ryota
Acta Crystallographica Section F, 61(8), p.788 - 790, 2005/08
Times Cited Count:7 Percentile:54.65(Biochemical Research Methods)Granulocyte colony-stimulating factor (GCSF) receptor receives signals for regulating the proliferation and differentiation of the precursor cells of granulocytes. The complex composed of two GCSFs and two GCSF receptors was crystallized. The crystal of the complex was grown in 1.0 M sodium formate and 0.1 M sodium acetate (pH4.6). It belongs to the space group 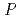 4
4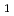 22 (or its enantiomorph 4
22 (or its enantiomorph 4 22) with unit cell dimensions of
22) with unit cell dimensions of 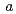 =
= 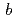 = 110.1 ,
= 110.1 , 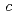 = 331.8 . However, the diffraction data from the crystal beyond 5 resolution could not be collected. Since the heterogeneity of GCSF receptor seems to interrupt growth of good quality crystals, the GCSF receptor was fractionated by achromatography. Crystals of GCSF/fractionated GCSF receptor complex were grown as a new crystal form in 0.2 M ammonium phosphate. The new crystal diffracts beyond 3.0 resolution and belongs to space group 321 (or its enantiomorph 3
= 331.8 . However, the diffraction data from the crystal beyond 5 resolution could not be collected. Since the heterogeneity of GCSF receptor seems to interrupt growth of good quality crystals, the GCSF receptor was fractionated by achromatography. Crystals of GCSF/fractionated GCSF receptor complex were grown as a new crystal form in 0.2 M ammonium phosphate. The new crystal diffracts beyond 3.0 resolution and belongs to space group 321 (or its enantiomorph 3 21) with unit-cell parameters = = 134.8, = 105.7 .
21) with unit-cell parameters = = 134.8, = 105.7 .
1 chain and IL-4 receptor Honjo, Eijiro; Shoyama, Yoshinari; Tamada, Taro; Arima, Kazuhiko*; Kanaji, Sachiko*; Izuhara, Kenji*; Kuroki, Ryota
no journal, ,
The receptor binding to Interleukin (IL)-13 is composed of the IL-13 receptor 1 chain (IL-13R1) and the IL-4 receptor chain (IL-4R). In order to investigate the interaction of IL-13 with IL-13R1 and IL-4R, the DNA fragments coding the extracellular regions of human IL-13R1 and the IL-4R, containing a cytokine receptor homologous region, were fused with human Fc and expressed by silkworm-baculovirus system. Size exclusion chromatography did not reveal association between IL-13 and IL-13R1 under these conditions, but the clear association of IL-13 and IL-13R1 was observed after adding IL-4R. This indicates that both extracellular regions of IL-13R1 and IL-4R have functions, and the association between IL-13 and IL-4R increases the IL-13 affinity to IL-13R1.
Honjo, Eijiro; Adachi, Motoyasu; Tamada, Taro; Kuroki, Ryota
no journal, ,
Because human immunodeficiency virus type 1 protease (HIV-1PR, 99 amino acids) is involved in the maturation of HIV-1, it is a prime target for antiviral therapy of AIDS. In order to investigate precise structure-function relationship, we are planning to determine the structure of HIV-1PR including the information of hydrogen and hydrating water molecules using high resolution X-ray crystallography and neutron crystallography. For higher resolution neutron protein crystallography, it is necessary to exchange hydrogen (H) atoms with deuterium (D) atoms in order to reduce background noise derived from incoherent neutron scattering cross-section of hydrogen. Therefore, we have expressed fully deuterated HIV-1PR using commercially available perdeuterated medium. X-ray structures of non labeled and perdeuterated HIV-1PR were determined to 1.2 and 1.4 resolution, respectively, using crystals grown under the same conditions. Both structures did not show any significant changes.
Honjo, Eijiro
no journal, ,
Perdeuteration of proteins was one of the important techniques to collect high resolution diffraction data in neutron protein crystallography. We have improved the preparation method of perdeuterated protein using HIV protease as one of the target proteins. Here, we report the refined protocols for cultivation of E. coli expressing HIV protease and refolding of HIV protease from its inclusion bodies. In addition, structural comparison between native hydrogenated and perdeuterated HIV protease will be mentioned.
Kuroki, Ryota; Adachi, Motoyasu; Honjo, Eijiro; Kurihara, Kazuo; Ohara, Takashi; Tamada, Taro
no journal, ,
Determination of hydrogens by neutrons diffraction enables us to observe the ionization status of amino acids and the structural features of hydrogen bonds and hydrating waters. It is, however, needed to obtain a huge protein crystal because the intencity of neutron beam is weak in conparison with X-ray beam. Contribution of neutron crystallography has been limited because it needed more than 2mm volume crystal and long term of data collection. Recent development on the palse sparation sourse is suposed to improve the efficiency of data collection more than 50 times, but necessity of a huge crystal still remains. In order to improve the size of the crystal, we have been working on the crystal lattice engineering to improve the volume of the crystal as well as the preparation of fully deuterated sample of proteins.
volume crystal and long term of data collection. Recent development on the palse sparation sourse is suposed to improve the efficiency of data collection more than 50 times, but necessity of a huge crystal still remains. In order to improve the size of the crystal, we have been working on the crystal lattice engineering to improve the volume of the crystal as well as the preparation of fully deuterated sample of proteins.
Niimura, Nobuo*; Tanaka, Ichiro*; Onishi, Yuki*; Ostermann, A.*; Kurihara, Kazuo; Honjo, Eijiro; Kuroki, Ryota; Futami, Junichiro*; Yamada, Hidenori*
no journal, ,
Our goal is to develop new methods for determining macromolecular crystal structures  from neutron diffraction data and to validate these methods by making applications to several proteins. The proposed methods, which have the potential to be more powerful than X-ray ones, exploit perfectly isomorphous pairs of crystals that differ by replacement of D atoms with H atoms in selected amino-acid residues. This research involves collaboration among teams on three continents that have expertise in different aspects of neutron crystallography. The Japanese team measures high-resolution neutron diffraction data from protein crystals to answer questions about H bonding, protonation, hydration, and enzyme mechanisms. The team will focus on fundamental and simple proteins such as myoglobin, insulin, and RNase A at the first stage and then on rubredoxin, aldose reductase and other samples from the French team. In the meeting the research plan of the Japanese team will be reported.
from neutron diffraction data and to validate these methods by making applications to several proteins. The proposed methods, which have the potential to be more powerful than X-ray ones, exploit perfectly isomorphous pairs of crystals that differ by replacement of D atoms with H atoms in selected amino-acid residues. This research involves collaboration among teams on three continents that have expertise in different aspects of neutron crystallography. The Japanese team measures high-resolution neutron diffraction data from protein crystals to answer questions about H bonding, protonation, hydration, and enzyme mechanisms. The team will focus on fundamental and simple proteins such as myoglobin, insulin, and RNase A at the first stage and then on rubredoxin, aldose reductase and other samples from the French team. In the meeting the research plan of the Japanese team will be reported.
Kuroki, Ryota; Honjo, Eijiro; Tamada, Taro
no journal, ,
Silk worm expresssion system is the rapid system for several protein expressions developed by Katakura Industries company. We have used this system for test expression of the extracellular region of receptors. Every cDNA coding extracellular region of receptors were fused to the cDNA of Fc region of antibody, and cloned into the transfer vector and the vector was transformed into vacuro virus that can infect silk worm. So far, expression of 10 extracellular regions of menbrane proteins were tested using this system, all proteins were successfully expressed. Among these proteins, interleukin-13 receptor alpha 1 and 2, interluekin-4 receptor were purified from the silk worm and their functions were comfirmed.
Adachi, Motoyasu; Honjo, Eijiro; Tamada, Taro; Blaber, M.*; Kuroki, Ryota
no journal, ,
Human acidic fibroblast growth factor (haFGF) is a powerful mitogen and angiogenic factor. Our objective is to reveal structure function relationships of haFGF by protein neutron crystallography. For the determination of neutron crystal structures, large crystal in size is required. Since haFGF forms thin crystals, we tried improvement of crystal growth by changing amino acids at molecular interface. The analysis using X-ray crystal structure showed that the residue of Glu81 is located near Glu81 generated by crystallographic symmetry. Therefore, Glu81 was replaced with Ala, Val, Leu, Ser and Thr residue. The mutants of E81S and E81T provided larger crystal in size than that of WT. The X-ray structures of E81S and E81T showed that a molecule of formic acid bridges between the two Glu81 residues by hydrogen bonding.
1 chain and its interaction with its ligandKuroki, Ryota; Honjo, Eijiro; Tamada, Taro; Arima, Kazuhiko*; Izuhara, Kenji*
no journal, ,
Interleukin-13 (IL-13) interacts with two types of receptors, the IL-13R1 chain (IL-13R1) and the IL-4R chain (IL-4R), that transduce the IL-13 signals. In order to investigate the interaction of IL-13 with IL-13R1 and IL-4R, the genes coding the extracellular regions of human IL-13R1 and the IL-4R containing cytokine receptor homologous region (CRH), were fused with human Fc, and expressed by silk worm. After further purification with anion-exchange chromatography, the receptors were used to investigate the ligand-receptor interaction. The size exclusion chromatography showed that association between IL-13 and IL-13Ra1 could not be observed in this condition whereas the clear association of IL-13 and IL-13Ra1 was observed after adding IL-4R. It indicates that the association between IL-13 and IL-4R increase the IL-13 affinity to IL-13R1.
