


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Itakura, Mitsuhiro; Kaburaki, Hideo; Yamaguchi, Masatake; Tsuru, Tomohito
Physical Review Letters, 116(22), p.225501_1 - 225501_5, 2016/06
Times Cited Count:40 Percentile:85.16(Physics, Multidisciplinary)Dislocations in close packed metals usually dissociate into a planar shape and their slip is confined in the corresponding slip planes. Cross-slip usually requires transformation of the planar dislocation core into a perfect dislocations, which requires high activation energy. Using an extensive DFT calculations, we have found a notable exception to this conventional view. The pyramidal 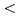 c+a
c+a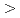 screw dislocation in Mg consists of two partial dislocations connected by a stacking fault, and the stacking fault can migrate perpendicular to the plane by atom shuffling, enabling the dislocation to cross-slip without transforming into a perfect dislocation.
screw dislocation in Mg consists of two partial dislocations connected by a stacking fault, and the stacking fault can migrate perpendicular to the plane by atom shuffling, enabling the dislocation to cross-slip without transforming into a perfect dislocation.
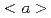 dislocation in magnesium
dislocation in magnesiumItakura, Mitsuhiro; Kaburaki, Hideo; Yamaguchi, Masatake; Tsuru, Tomohito
Modelling and Simulation in Materials Science and Engineering, 23(6), p.065002_1 - 065002_19, 2015/09
Times Cited Count:15 Percentile:51.61(Materials Science, Multidisciplinary)The cross-slip process of a screw dislocation from the basal to the prismatic plane in magnesium was studied using the density functional theory and the molecular dynamics calculations. An atomistic method for calculating the total Peierls energy map has been devised to track the transition path of a dissociated and/or constricted screw dislocation in the cross-slip process. The barrier of a screw dislocation from the basal to the prismatic plane is estimated by the density functional theory for the first time. The activation enthalpy for the cross slip is calculated using a line tension model based on the density functional theory to be 1.4 to 1.7 eV, which is in reasonable agreement with experiments. On the basis of the results, the effect of temperature on the cross-slip process of the dissociated screw dislocation on the basal plane is studied in detail using the molecular dynamics method.
Ebihara, Kenichi; Kaburaki, Hideo; Itakura, Mitsuhiro
"Hagane No Kikaiteki Tokusei Ni Oyobosu Suiso No Koka To Sono Hyoka" Shimpojium Yokoshu (USB Flash Drive), 6 Pages, 2014/09
Since hydrogen(H) embrittlement is one factor causing degradation and/or fracture of steel, understanding its mechanism is required. The grain-boundary(GB) decohesion due to segregation of H is considered to cause the delayed fracture of high strength steels and the cold cracking in welding. In the model based on GB decohesion, information of strength of GBs estimated in the atomic scale is used for the estimation of strength or crack propagation in the macroscopic scale. However the modeling between the atomic and the macroscopic scales is not clear. In particular, the validity of the model using the elastic continuum around nano-cracks for stress concentration at the crack tip is not clear. Thus, we examined the difference of the stress distribution around the nano-crack which was estimated by molecular dynamics and by a continuum calculation. As a result, the discrepancy became remarkable at high strain. The stress concentration was not simulated by the elastic continuum model.
Ebihara, Kenichi; Kaburaki, Hideo; Takai, Kenichi*
Zairyo To Purosesu (CD-ROM), 27(1), P. 418, 2014/03
In order to understand the mechanism of hydrogen embrittlement, identifying the state of hydrogen trapped by defects in steels is dispensable. In the identification of the hydrogen trapping state, thermal desorption profiles of hydrogen obtained in the thermal desorption analysis of steel specimens are widely used, and need to be analyzed using the numerical model because they include the effect of the specimen size and the experimental conditions as well as the effect of defects. The prefactor of detrap rate in the model is previously used as a fitting parameter. In the presentation, the influence of specimen size on the identification of the prefactor was examined numerically. As a result, in the specimens of pure iron and martensite steels whose size is larger than 0.3 mm, the accuracy of the identification rapidly drops. In addition, according to the influence of the prefactor on the desorption profile, it is possible to identify the order of magnitude of the prefactor.
Ebihara, Kenichi; Kaburaki, Hideo; Takai, Kenichi*
Proceedings of 2012 International Hydrogen Conference; Hydrogen-Materials Interactions, p.553 - 561, 2014/02
The crack causing hydrogen embrittlement is observed in steels as the structural material. Accurate evaluation of hydrogen detrapping activation energy, which represents the binding strength between hydrogen atoms and the lattice defects, is crucial to the understanding of the mechanism of hydrogen embrittlement in steels. The Choo and Lee's method, which experimentally evaluates the detrapping energy from the hydrogen thermal desorption profile, should be scrutinized, because this method neglects the hydrogen diffusion in the specimen. By their method, we have evaluated detrapping activation energies from the experimental desorption profiles for pure iron, and also from that simulated by the 1D reaction-diffusion equation. We found that their method underestimates the detrapping energies as the specimen size is large. We also found that this dependence on the specimen size is caused by the degradation of the desorption peak of the detrapping process by the diffusion process.
Suzudo, Tomoaki; Tsuru, Tomohito; Yamaguchi, Masatake; Kaburaki, Hideo
Journal of Nuclear Materials, 442(1-3), p.S655 - S659, 2013/11
Times Cited Count:7 Percentile:46.40(Materials Science, Multidisciplinary)He bubbles at grain boundaries (GBs) may lead materials to serious embrittlement, but the mechanism of the nucleation of such bubbles is not well-understood. In the present paper, we analyzed the stability of various He-vacancy clusters, which are precursors of He bubbles, at several kinds of grain boundaries of alpha-Fe using a set of empirical potentials. We found that the dissociation energy of vacancy from He-vacancy clusters at GBs is generally less than that for the intragranular counterparts, and that the He-to-vacancy ratio in equilibrium for various GB cases becomes larger in comparison with the intragranular case. The results obtained here are useful for kinetic models of He bubble nucleation.
Itakura, Mitsuhiro; Kaburaki, Hideo; Yamaguchi, Masatake; Okita, Taira*
Acta Materialia, 61(18), p.6857 - 6867, 2013/10
Times Cited Count:154 Percentile:98.73(Materials Science, Multidisciplinary)The interaction between dislocations and impurity atoms in metals determines various properties of plastic deformation, such as the dependence of the yield stress on the impurity contents. Since the direct observation of atomistic structure of screw dislocation is almost impossible, several hypothetical assumptions have been employed to explain conveniently experimental observations. Recent advancement of computational hardware, as well as the development of elaborated techniques to reduce the size-effect in the first-principles calculation, enabled direct calculations of dislocation-impurity interaction. We have succeeded to evaluate the effect of hydrogen atoms on the dislocation mobility in iron.
Tsuru, Tomohito; Udagawa, Yutaka; Yamaguchi, Masatake; Itakura, Mitsuhiro; Kaburaki, Hideo; Kaji, Yoshiyuki
Journal of Physics; Condensed Matter, 25(2), p.022202_1 - 022202_5, 2013/01
Times Cited Count:57 Percentile:85.67(Physics, Condensed Matter)There is a pressing need to improve the ductility of magnesium alloy toward the advanced application for light-weight structural materials. In this letter we focused attention on the particular potential function for yttrium to activate the non-basal slip. The generalized stacking fault (GSF) energies of both basal and prismatic planes for pure magnesium are calculated by density functional theory (DFT) and EAM potential, and its effect on the dislocation core structures are examined by semidiscrete variation Peierls Nabarro (SVPN) model. Solution softening of added yttrium was similarly estimated by combination of DFT and SVPN model. Yttrium was found to have a particular influence on solution softening by the reduction of the gradient of the GSF energy.
Ebihara, Kenichi; Kaburaki, Hideo
Suiso Zeika Kenkyu No Kiban Kochiku Chukan Hokokukai Yokoshu, p.27 - 34, 2012/09
The thermal desorption analysis (TDA) is the experimental method to identify hydrogen state, which is necessary for understanding the mechanism of hydrogen embrittlement in steels. From the thermal desorption profile obtained by TDA, the binding energy between hydrogen and defects, which is important for estimating the hydrogen state, can be calculated. Since the desorption profile is influenced by the specimen size and the initial hydrogen state before starting TDA, the understanding of their influence on the desorption profile is necessary. By simulating the desorption profile using the reaction-diffusion equations incorporating the parameters which were corrected by reproducing the experimental profile of martensitic steels, we confirmed that the profile peak becomes broad and the calculated binding energy becomes low when the size is large, and found that two desorption peaks are observed when the non-equilibrium state is used as initial state for the large-size specimen.
Itakura, Mitsuhiro; Kaburaki, Hideo; Yamaguchi, Masatake
Acta Materialia, 60(9), p.3698 - 3710, 2012/05
Times Cited Count:97 Percentile:96.07(Materials Science, Multidisciplinary)Irradiation hardening of nuclear materials are caused by lattice defects which hinder the motion of dislocations and thus suppress plastic deformations. To understand the irradiation hardening, the precise knowledge about the dislocation motion inside the material is indispensable. In bcc metals the mobility of dislocations is determined by the atomic structure of the dislocation and thus quantum mechanical calculation is required to estimate the mobility of dislocations. We have devised a new method to calculate the dislocation properties combining linear elasticity theory and quantum mechanical calculations, and also developed a new method to control the two-dimensional motion of dislocation in the simulation. These new methods allowed us to identify the reason why wrong dislocation motion is observed in the current molecular dynamics simulations, and to present a guideline to improve these simulations. This work opened a way for the quantitative simulations of irradiation hardening.
Yamaguchi, Masatake; Kameda, Jun*; Ebihara, Kenichi; Itakura, Mitsuhiro; Kaburaki, Hideo
Philosophical Magazine, 92(11), p.1349 - 1368, 2012/04
Times Cited Count:69 Percentile:92.35(Materials Science, Multidisciplinary)Atomistic mechanisms of hydrogen-induced cracking along a bcc Fe 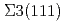 symmetrical tilt grain boundary (GB) have been studied by first-principles calculations. The mobile and immobile effects of hydrogen on the GB decohesion are analyzed by calculating the dependence of hydrogen segregation energy on the coverage relevant to the repulsive interaction among segregated hydrogen atoms at the GB and on its fracture surfaces, together with generalizing McLean's formula. It was found that the segregation of combined mobile and immobile hydrogen atoms from the bulk and/or GB on the fracture surfaces causes much stronger reduction (70-80%) in the GB cohesive energy. It can occur even at a very low bulk hydrogen content of about 10
symmetrical tilt grain boundary (GB) have been studied by first-principles calculations. The mobile and immobile effects of hydrogen on the GB decohesion are analyzed by calculating the dependence of hydrogen segregation energy on the coverage relevant to the repulsive interaction among segregated hydrogen atoms at the GB and on its fracture surfaces, together with generalizing McLean's formula. It was found that the segregation of combined mobile and immobile hydrogen atoms from the bulk and/or GB on the fracture surfaces causes much stronger reduction (70-80%) in the GB cohesive energy. It can occur even at a very low bulk hydrogen content of about 10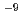 atomic fraction during slow cracking. This is in contrast with only 10-20% decohesion induced by immobile hydrogen at much higher hydrogen content during fast cracking.
atomic fraction during slow cracking. This is in contrast with only 10-20% decohesion induced by immobile hydrogen at much higher hydrogen content during fast cracking.
Ebihara, Kenichi; Kaburaki, Hideo
ISIJ International, 52(2), p.181 - 186, 2012/02
Times Cited Count:19 Percentile:36.90(Metallurgy & Metallurgical Engineering)In the study of hydrogen embrittlement, it is indispensable to identify the hydrogen trapping state in steels. The hydrogen thermal desorption profile, which is obtained from the thermal desorption analysis (TDA) and is the relation between temperature and the amount of desorbed hydrogen of a specimen heated at a constant rate, is effective. Since various factors such as the specimen size, the heating rate, the hydrogen diffusion and the trapping effect of defects affect the desorption profile, however, it is necessary to model the process of the hydrogen desorption from the specimen. This paper reviewed the present model which can simulate the desorption profile by categorizing them according to the rate-determining process of the hydrogen desorption. The historical background and the range of validity of the models were also mentioned.
 -Fe; Damage energy and temperature dependence analyses
-Fe; Damage energy and temperature dependence analysesSuzudo, Tomoaki; Golubov, S. I.*; Stoller, R. E.*; Yamaguchi, Masatake; Tsuru, Tomohito; Kaburaki, Hideo
Journal of Nuclear Materials, 423(1-3), p.40 - 46, 2012/01
Times Cited Count:8 Percentile:50.20(Materials Science, Multidisciplinary)In this paper, kinetic Monte Carlo method was applied to investigate the long time evolution of cascade damage prepared by molecular dynamics simulations in -Fe up to recoil energy of more than 200 keV. We conducted thorough investigation on how the surviving defects vary with cascade damage energy and annealing temperature. The results can be used for input parameters of rate equations to simulate microstructural evolution under irradiation. The study also suggested that neighboring sub-cascades evolves almost independently during annealing, and that the temperature dependence of the annealing results can be explained by the temperature dependence of vacancy-migration and vacancy-dissociation probabilities.
-FeSuzudo, Tomoaki; Kaburaki, Hideo; Yamaguchi, Masatake
Journal of Nuclear Materials, 417(1-3), p.1102 - 1105, 2011/10
Times Cited Count:12 Percentile:64.62(Materials Science, Multidisciplinary)This paper discusses a computational approach to the helium segregation at grain boundaries with reference to the helium embrittlement under the fast neutron irradiation; we particularly applied a kinetic Monte-Carlo method to the modeling of the segregation phenomena in alpha-iron. In the calculation, the migration and segregation energies of helium atoms given by the first principle calculations are used. Obtained results are discussed in comparison with McLean's equilibrium segregation theory.
Ebihara, Kenichi; Itakura, Mitsuhiro; Yamaguchi, Masatake; Kaburaki, Hideo; Suzudo, Tomoaki
Progress in Nuclear Science and Technology (Internet), 2, p.38 - 43, 2011/10
The decohesion model in which hydrogen segregating at grain boundaries reduces cohesive energy is considered to explain hydrogen embrittlement. In order to verify the decohesion model, it is necessary to evaluate stress and hydrogen concentration at grain boundaries under experimental conditions. Thus, we calculated the stress and the hydrogen concentration at grain boundaries in the 3-dimensional polycrystalline model generated by the random Voronoi tessellation. The crystallographic anisotropy was given to each grain as a characteristic. As the boundary conditions, data extracted from the results calculated in the notched round-bar specimen model under the tensile test condition was given to the polycrystalline model. As a result, it was found that the evaluated stress does not reach the fracture stress estimated by first-principles calculations. Therefore, it was considered that the initiation of grain boundary fracture needs some factors except the crystallographic anisotropy.
Yamaguchi, Masatake; Ebihara, Kenichi; Itakura, Mitsuhiro; Kadoyoshi, Tomoko*; Suzudo, Tomoaki; Kaburaki, Hideo
Metallurgical and Materials Transactions A, 42(2), p.330 - 339, 2011/02
Times Cited Count:139 Percentile:97.80(Materials Science, Multidisciplinary)Suzudo, Tomoaki; Yamaguchi, Masatake; Kaburaki, Hideo; Ebihara, Kenichi
Materials Research Society Symposium Proceedings, Vol.1215, 7 Pages, 2010/10
We applied ab initio calculation and an object kinetic Monte Carlo modeling to the study of He-vacancy cluster nucleation under irradiation in bcc and fcc Fe, which are surrogate materials for ferritic/martensitic and austenitic steels, respectively. The ab initio calculations provided parameters for the object kinetic Monte Carlo model, such as the migration energies of point defects and the dissociation energies of He and vacancy to He-vacancy clusters. We specially focused on the simulation of high He/dpa irradiation such as He-implantation into the materials and tracked the nucleation of clusters and the fate of point defects such as SIAs, vacancies, and He atoms. We found no major difference of He-vacancy cluster nucleation between bcc and fcc Fe when we ignore the intracascade clustering even if the migration energies of point defects are significantly different between the two crystals.
Ebihara, Kenichi; Itakura, Mitsuhiro; Yamaguchi, Masatake; Kaburaki, Hideo; Suzudo, Tomoaki
Proceedings of Joint International Conference of 7th Supercomputing in Nuclear Application and 3rd Monte Carlo (SNA + MC 2010) (USB Flash Drive), 6 Pages, 2010/10
The decohesion model in which hydrogen segregating at grain boundaries reduces their strength is considered to explain hydrogen embrittlement of steels. Therefore in order to understand hydrogen embrittlement from this model, stress and hydrogen concentration at grain boundaries need be evaluated under the fracture condition for tensile test specimens. From this consideration, we evaluated the stress and the hydrogen concentration at grain boundaries in the three-dimensional polycrystalline model which was generated by Voronoi tessellation. The different crystallographic orientation was given to each grain. Extracted data from the calculation in the notched round-bar specimen model under the tensile test condition was given to the polycrystalline model as the boundary condition. As a result, it was found that the valuated stress does not reach the fracture stress which was estimated under the condition of the evaluated hydrogen concentration by first principles calculation.
Yamaguchi, Masatake; Ebihara, Kenichi; Itakura, Mitsuhiro; Suzudo, Tomoaki; Kaburaki, Hideo
Proceedings of Joint International Conference of 7th Supercomputing in Nuclear Application and 3rd Monte Carlo (SNA + MC 2010) (USB Flash Drive), 4 Pages, 2010/10
It is not known in detail how much solute atoms segregate in grain boundaries of metals and how much the cohesive energy (work of fracture) of grain boundary is decreased by the segregation. From first-principles, we calculated the segregation energy of some solute elements like boron (B), carbon (C), phosphorous (P), and sulfur (S) in bcc Fe Sigma 3 (111) symmetrical tilt grain boundary with varying the segregation density. We find that these elements can segregate up to a high concentration in the grain boundary. We also find that the segregation energy on the fracture surface is significantly larger than that in the grain boundary for the embrittling elements like P and S. On the contrary, the cohesive energy is increased by B and C segregation. The increase-decrease rate in the calculated cohesive energy by solute segregation is found to be well correlated with experimentally observed shift in ductile-to-brittle transition temperature by solute segregation.
-FeSuzudo, Tomoaki; Golubov, S.*; Stoller, R.*; Yamaguchi, Masatake; Tsuru, Tomohito; Kaburaki, Hideo
Proceedings of Joint International Conference of 7th Supercomputing in Nuclear Application and 3rd Monte Carlo (SNA + MC 2010) (USB Flash Drive), 6 Pages, 2010/10
Molecular dynamics is a useful tool to analyze cascade damage in structural materials of nuclear devices, but the time scale accessible to molecular dynamics is 100 ps. Kinetic Monte Carlo annealing simulation of cascade damage is useful for analyzing the longer time development of cascade damage. We conducted a series of such annealing simulations in -Fe. The surviving displacement ratio to the NRT displacements before annealing is 0.3 in the case of primary knock-on atom's energy more than 10 keV, but it decreased by 30 % through the annealing at 300 K because of recombination of vacancies and self-interstitial atoms, and the recombination ratio increased as the annealing temperature increased. These results are meaningful when applied to the simulation of accumulation of cascades using rate theory. This work is useful for R&D of nuclear materials.
