


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Hou, Z.*; Wang, X.*; Ikeda, Takashi; Terakura, Kiyoyuki*; Oshima, Masaharu*; Kakimoto, Masaaki*
Physical Review B, 87(16), p.165401_1 - 165401_16, 2013/04
Times Cited Count:115 Percentile:95.34(Materials Science, Multidisciplinary)We have performed the DFT calculations to study the electronic structures of N-doped graphene with vacancies and Stone-Wales defect. Our results show that monovacancies in graphene act as hole dopants and that two substitutional N dopants are needed to compensate for the hole introduced by a monovacancy. On the other hand, divacancy does not produce any free carriers. Interestingly, a single N dopant at divacancy acts as an acceptor rather than a donor. Compared with the case of an isolated N dopant in perfect graphene, the electrons donated by substitutional N dopants would be localized significantly when a N-N pair is formed. The N-N interaction and the interference between native point defect and N dopant strongly modify the role of N doping regarding the free carrier production in the bulk 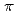 bands. Our results are qualitatively consistent with the experimental observation that the concentration of free electrons introduced by N dopants would be lower than that of doped N.
bands. Our results are qualitatively consistent with the experimental observation that the concentration of free electrons introduced by N dopants would be lower than that of doped N.
Wang, X.*; Hou, Z.*; Ikeda, Takashi; Oshima, Masaharu*; Kakimoto, Masaaki*; Terakura, Kiyoyuki*
Journal of Physical Chemistry A, 117(3), p.579 - 589, 2013/01
Times Cited Count:40 Percentile:80.62(Chemistry, Physical)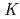 -edge X-ray absorption (XAS), emission (XES), and photoelectron (XPS) spectra of nitrogen doped along graphene edges are systematically investigated by using first-principles methods. In this study we considered pyridinium-like, pyridine-like, cyanide-like, and aminelike nitrogens at armchair and zigzag edges and pyrrole-like nitrogen at armchair edge as well as graphite-like nitrogen at graphene interior site. Our results indicate that nitrogen configuration and its location (armchair or zigzag edge) in nitrogen-doped graphene can be identified via the spectral analysis. Furthermore, some controversial spectral features observed in experiment for N-doped graphene-like materials are unambiguously assigned. The present analysis gives an explanation to the reason why the peak assignment is usually made differently between XPS and XAS.
-edge X-ray absorption (XAS), emission (XES), and photoelectron (XPS) spectra of nitrogen doped along graphene edges are systematically investigated by using first-principles methods. In this study we considered pyridinium-like, pyridine-like, cyanide-like, and aminelike nitrogens at armchair and zigzag edges and pyrrole-like nitrogen at armchair edge as well as graphite-like nitrogen at graphene interior site. Our results indicate that nitrogen configuration and its location (armchair or zigzag edge) in nitrogen-doped graphene can be identified via the spectral analysis. Furthermore, some controversial spectral features observed in experiment for N-doped graphene-like materials are unambiguously assigned. The present analysis gives an explanation to the reason why the peak assignment is usually made differently between XPS and XAS.
Kiuchi, Hisao*; Niwa, Hideharu*; Kobayashi, Masaki*; Harada, Yoshihisa*; Oshima, Masaharu*; Chokai, Masayuki*; Nabae, Yuta*; Kuroki, Shigeki*; Kakimoto, Masaaki*; Ikeda, Takashi; et al.
Electrochimica Acta, 82(1), p.291 - 295, 2012/10
Times Cited Count:14 Percentile:32.74(Electrochemistry)We study the characteristics of oxygen adsorption on metal-free carbon-based cathode catalysts derived from nitrogen-containing polyamide (PA) and nitrogen-free phenolic resin (PhRs). Electrochemical analysis and Raman spectroscopy showed higher 2-electron oxygen reduction reaction (ORR) activity and more defect sites in PA than PhRs. The increase in the amount of adsorbed oxygen in PA was also identified by oxygen adsorption isotherms. 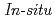 X-ray photoelectron spectroscopy reveals that graphite-like nitrogen contributes to oxygen adsorption and C=O components are dominant in PA. These experimental results indicate that the adsorbed C=O components near the graphite-like nitrogen can be assigned as active sites for 2-electron ORR.
X-ray photoelectron spectroscopy reveals that graphite-like nitrogen contributes to oxygen adsorption and C=O components are dominant in PA. These experimental results indicate that the adsorbed C=O components near the graphite-like nitrogen can be assigned as active sites for 2-electron ORR.
Hou, Z.*; Wang, X.*; Ikeda, Takashi; Terakura, Kiyoyuki*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
Physical Review B, 85(16), p.165439_1 - 165439_9, 2012/04
Times Cited Count:138 Percentile:96.32(Materials Science, Multidisciplinary)To understand the interaction between nitrogen dopants and point defects in graphene, we have studied energetic stability of N-doped graphene with vacancies and Stone-Wales defect by performing the density functional theory calculations. Our results show that N substitution energetically prefers to occur at the carbon atoms near the defects, especially for those sites with larger bond shortening, indicating that the defect-induced strain plays an important role in the stability of N dopants. In the presence of mono-vacancy, the most stable position for N is the pyridine-like configuration while for other point defects studied N prefers a site in the pentagonal ring. While the N doping is endothermic in defect-free graphene, it becomes exothermic for defective one. Our results imply that the point defect and N dopant attract each other, which means that substitutional N dopants would increase the probability of point defect generation and vice versa.
Wang, X.*; Hou, Z.*; Ikeda, Takashi; Huang, S.-F.*; Terakura, Kiyoyuki*; Boero, M.*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
Physical Review B, 84(24), p.245434_1 - 245434_7, 2011/12
Times Cited Count:33 Percentile:74.81(Materials Science, Multidisciplinary)The structural and electronic properties of N-doped zigzag graphene ribbons with various ratios of di- to monohydrogenated edge carbons are investigated within the density functional theory framework. We find that the stability of graphitic N next to the edge, which is claimed to play important roles in the catalytic activity in our previous work, will be enhanced with increasing the concentration of di-hydrogenated carbons. Furthermore, the di-hydrogenated edge carbons turn out to be easily converted into mono-hydrogenated ones in the presence of oxygen molecules at room temperature. Based on our results, we propose a possible way to enhance the oxygen reduction catalytic activity of N-doped graphene by controlling the degrees of hydrogenation of edge carbons. The characteristic features in the X-ray absorption and emission spectra for each specific N site considered here will also be given.
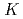 -edge X-ray absorption spectra of nanographene
-edge X-ray absorption spectra of nanographeneHou, Z.*; Wang, X.*; Ikeda, Takashi; Huang, S.-F.*; Terakura, Kiyoyuki*; Boero, M.*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
Journal of Physical Chemistry C, 115(13), p.5392 - 5403, 2011/03
Times Cited Count:38 Percentile:70.69(Chemistry, Physical)Carbon -edge X-ray absorption spectra of nanographene have been simulated by density functional theory calculations in order to obtain the information on the edge termination by hydrogen. Our results show that different edge terminations significantly affect the binding energy of 1s core-level of C atoms in the vicinity of edges because of the change in chemical bonding and the localized edge states. We find that a shoulder or a peak appears below the 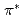 peak at relatively different positions with respect to the peak position in the theoretical spectra of zigzag graphene nano-ribbons, depending on the ratio of mono-hydrogen- to di-hydrogen-terminations. We also point out that the two additional features observed between the and
peak at relatively different positions with respect to the peak position in the theoretical spectra of zigzag graphene nano-ribbons, depending on the ratio of mono-hydrogen- to di-hydrogen-terminations. We also point out that the two additional features observed between the and 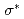 peaks of an ideal graphene originate from the states of C-H bonding and C-H
peaks of an ideal graphene originate from the states of C-H bonding and C-H bonding at the edges.
bonding at the edges.
Ikeda, Takashi; Hou, Z.*; Wang, X.*; Huang, S.-F.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
no journal, ,
no abstracts in English
Ikeda, Takashi; Hou, Z.*; Wang, X.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
no journal, ,
no abstracts in English
Ikeda, Takashi; Hou, Z.*; Wang, X.*; Huang, S.-F.*; Terakura, Kiyoyuki*; Oshima, Masaharu*; Kakimoto, Masaaki*; Miyata, Seizo*
no journal, ,
no abstracts in English
