


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 O)
O)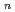 (
(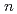 = 1-7) clusters on semi-empirical PM6 potential energy surfaces
= 1-7) clusters on semi-empirical PM6 potential energy surfacesYoshikawa, Takehiro*; Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemical Physics, 365(1-2), p.60 - 68, 2009/12
Times Cited Count:15 Percentile:44.84(Chemistry, Physical)Path-integral molecular dynamics simulations for the hydrogen-bonded glycine-(HO) ( = 1-7) clusters have been carried out using an on-the-fly direct dynamics technique at the semiempiricalPM6 level of theory. In the case of smaller clusters with = 1-3, the simulations show that the clusterstructure takes exclusively the hydrogen-bonded complex between a canonical neutral glycine and a water cluster moiety. In contrast, it was found that proton-exchange processes effectively occur between the COOH carboxylic group and water cluster moiety for = 4-6 clusters although the overall structures are the complex between a neutral glycine and water clusters. In the case of the = 7 cluster, glycine preferentially takes a zwitterionic form having NH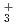 and COO
and COO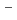 functional groups.
functional groups.
SO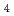
 (HO) (=1-6) on semiempirical PM6 potential surfaces
(HO) (=1-6) on semiempirical PM6 potential surfacesKakizaki, Akira*; Motegi, Haruki*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
Quantum path-integral molecular dynamics simulations for a series of sulfuric acid clusters, HSO(HO)(=1-6), were performed using semiempirical PM6 method. It was found that the acid dissociation probability increases with an increase in the cluster size, and that so-called contact-ion-pair structures are dominant in the proton-dissociated clusters. The importance of nuclear quantum effects in the cluster structures and proton-transfer processes is demonstrated.
O) on a semiempirical potential energy surfaceTakayanagi, Toshiyuki*; Takahashi, Kenta*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics, 358(3), p.196 - 202, 2009/04
Times Cited Count:16 Percentile:47.35(Chemistry, Physical)Path-integral molecular dynamics simulations for the HCl(HO) cluster have been performed on the ground-state potential energy surface directly obtained on-the-fly from semiempirical PM3-MAIS molecular orbital calculations. It is found that the HCl(HO) cluster has structural rearrangement above the temperature of 300 K showing a liquid-like behavior. Quantum mechanical fluctuation of hydrogen nuclei plays a significant role in structural arrangement processes in this cluster.
Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Taketsugu, Yuriko*; Taketsugu, Tetsuya*; Shiga, Motoyuki
Chemical Physics, 354(1-3), p.38 - 43, 2008/12
Times Cited Count:12 Percentile:37.98(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed to understand the quantum helium solvation structures in the He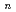 BeO cluster. Our simulations show that one helium atom is strongly bound to BeO to form HeBeO and that the first solvation shell around the HeBeO complex includes roughly 12-14 helium atoms. The present simulations implies that the HeBeO complex could be experimentally detected in large helium clusters using the HENDI technique.
BeO cluster. Our simulations show that one helium atom is strongly bound to BeO to form HeBeO and that the first solvation shell around the HeBeO complex includes roughly 12-14 helium atoms. The present simulations implies that the HeBeO complex could be experimentally detected in large helium clusters using the HENDI technique.
 O) (=2-7) clusters on semiempirical PM6 potential energy surfaces
O) (=2-7) clusters on semiempirical PM6 potential energy surfacesTakayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
Molecular dynamics simulations based on semiempirical PM6 method was carried out to study statistical structures of glycine-water clusters. We found that zwitterionic form is more stable than neutral form as the cluster size increases. The difference in water solvation structures around the neutral or zwitterionic glycine form is also discussed.
Takizawa, Daichi*; Takagi, Hitoshi*; Makita, Chikako*; Nakajima, Yuka*; Saito, Etsuko*; Oyama, Tatsuya*; Ichikawa, Takeshi*; Kakizaki, Akira*; Sato, Ken*; Mori, Masatomo*; et al.
no journal, ,
no abstracts in English
