


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 ) by CCSD and CCSD(T) calculations
) by CCSD and CCSD(T) calculationsNakazawa, Tetsuya
AIP Advances (Internet), 11(4), p.045032_1 - 045032_8, 2021/04
Times Cited Count:1 Percentile:4.48(Nanoscience & Nanotechnology)Geometries and energy separations of various low-lying electronic states of iron trimer (Fe) are investigated by CCSD and CCSD(T) calculations. The ground state is found to be a 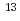 A' state with C
A' state with C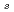 symmetry, whereas a nearly isoenergetic state, A
symmetry, whereas a nearly isoenergetic state, A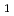 (C
(C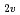 ), is degenerate to the ground state. The ground and five low-lying states with a spin multiplicity of 13 are found below 0.20 eV at the CCSD(T) level. On the other hand, the low-lying states with spin multiplicities of 9, 11, and 15 appear only above 0.20 eV. From detailed natural bond orbital (NBO) analyses, Fe has Fe-Fe bonds comprised of
), is degenerate to the ground state. The ground and five low-lying states with a spin multiplicity of 13 are found below 0.20 eV at the CCSD(T) level. On the other hand, the low-lying states with spin multiplicities of 9, 11, and 15 appear only above 0.20 eV. From detailed natural bond orbital (NBO) analyses, Fe has Fe-Fe bonds comprised of 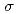 -bond orbitals only in
-bond orbitals only in 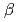 -spin part with higher s-character in low-lying states with a spin multiplicity of 13. The polarization coefficients indicate that the
-spin part with higher s-character in low-lying states with a spin multiplicity of 13. The polarization coefficients indicate that the 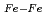 bonds are nearly complete covalent bonds with little polarization.
bonds are nearly complete covalent bonds with little polarization.
 O and the reaction of Fe + O
O and the reaction of Fe + ONakazawa, Tetsuya
Computational Materials Science, 146, p.334 - 345, 2018/04
Times Cited Count:2 Percentile:6.41(Materials Science, Multidisciplinary)Density functional theory calculations are performed on all possible structures of FeO using the hybrid B3LYP functional and the B3LYP functional combined with the broken-symmetry (BS) approach to obtain the most stable isomers. Based on the obtained stable isomers, the reaction mechanism of Fe + O toward rhombic Fe(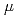 -O) is considered. The BS-singlet state of the rhombic Fe(-O) 2.1 is found to be the ground state of all FeO isomers. The
-O) is considered. The BS-singlet state of the rhombic Fe(-O) 2.1 is found to be the ground state of all FeO isomers. The 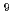 A" state of the open-cycle (
A" state of the open-cycle (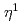 -O) Fe(-O) 2.11 and
-O) Fe(-O) 2.11 and  A state of the near-linear OFeOFe 1.4 are found to have the second and third lowest energy states. The lowest-lying energy states of the bare FeO clusters do not favor three-dimensional structures, but favor the linear and planar structures. The singlet and nonet energy surfaces calculated for the reaction of Fe + O toward rhombic Fe(-O) suggest that the reaction adiabatically proceeds with spin inversion from the nonet to singlet state.
A state of the near-linear OFeOFe 1.4 are found to have the second and third lowest energy states. The lowest-lying energy states of the bare FeO clusters do not favor three-dimensional structures, but favor the linear and planar structures. The singlet and nonet energy surfaces calculated for the reaction of Fe + O toward rhombic Fe(-O) suggest that the reaction adiabatically proceeds with spin inversion from the nonet to singlet state.
with ONakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 117, p.455 - 467, 2016/05
Times Cited Count:8 Percentile:26.08(Materials Science, Multidisciplinary)The reactions of Fe and FeO with O and the products of these reactions are investigated at the B3LYP/6-311+G(d) level. The reactions are considered in terms of the calculated potential energy surfaces, the interaction energies between reactant species, and the energies required to populate the higher electronic states such as the excited states of Fe(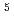 F, F) and O(
F, F) and O(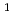
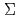
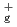 ). It is found that the reactions of Fe with O are endothermic and that the direct formation of dioxide OFeO is due to an ionic interaction of Fe
). It is found that the reactions of Fe with O are endothermic and that the direct formation of dioxide OFeO is due to an ionic interaction of Fe with O
with O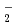 . Furthermore, the diabatic transitions from the covalent and ionic surfaces onto another ionic surface with energy barriers allow the Fe + O reaction to proceed toward the formation of OFeO. There are other paths corresponding to the vertical excitation from peroxide Fe(O) to dioxide OFeO. The OFeO + O reactions are found to endothermically produce the
. Furthermore, the diabatic transitions from the covalent and ionic surfaces onto another ionic surface with energy barriers allow the Fe + O reaction to proceed toward the formation of OFeO. There are other paths corresponding to the vertical excitation from peroxide Fe(O) to dioxide OFeO. The OFeO + O reactions are found to endothermically produce the 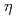 2- and -(O)FeO complexes, and the low-lying states of complexes are found to be closely located in energy. The CCSD and CCSD(T) single point energy calculations are performed with the structures optimized at the B3LYP level. The results of NBO and Mulliken analyses and the harmonic vibrational frequencies are presented.
2- and -(O)FeO complexes, and the low-lying states of complexes are found to be closely located in energy. The CCSD and CCSD(T) single point energy calculations are performed with the structures optimized at the B3LYP level. The results of NBO and Mulliken analyses and the harmonic vibrational frequencies are presented.
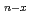 Si
Si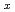 (
(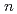 ,
, 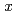
 6) clusters; Theoretical investigation based on density functional theory
6) clusters; Theoretical investigation based on density functional theoryNakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 68, p.350 - 360, 2013/02
Times Cited Count:4 Percentile:14.17(Materials Science, Multidisciplinary)Based on DFT calculations, FeSi (, 6) clusters are investigated. The Si substitutions increase cohesion of Fe cluster. The substitutions affect structural parameter, bond strength, and magnetic moment. The changes are connected with electron transfer from Fe to Si atoms.
Mo (, 6) clustersNakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 55, p.365 - 375, 2012/04
Times Cited Count:12 Percentile:35.44(Materials Science, Multidisciplinary)Igarashi, Takahiro; Nakazawa, Tetsuya; Suzuki, Chikashi; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 50(12), p.3346 - 3349, 2011/12
Times Cited Count:1 Percentile:4.27(Materials Science, Multidisciplinary)The authors have developed a new approach for large-scale systems including over 100,000 atoms to obtain physical strength from the viewpoint of atom-atom bonding energy. In this study, the authors applied this method to SiH molecule and crystalline silica system, and carried out bond order and bonding energy analyses. In this analysis, the developed method offered almost the same analytical accuracy as the first principle method, while its calculation speed was much faster than that of the latter.
molecule and crystalline silica system, and carried out bond order and bonding energy analyses. In this analysis, the developed method offered almost the same analytical accuracy as the first principle method, while its calculation speed was much faster than that of the latter.
Nakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki; Jitsukawa, Shiro
Journal of Nuclear Materials, 417(1-3), p.1090 - 1093, 2011/10
Times Cited Count:4 Percentile:31.12(Materials Science, Multidisciplinary)It is well known that impurities in iron which segregate to grain boundaries can dramatically change physical and chemical properties. Phosphorous, which is one of impurities, segregates at grain boundaries under thermal or irradiation environments, and brings about the intergranual embrittlement. In this study, influences of phosphorus substitutions for binding energies and electronic structures of octahedral iron cluster are investigated computationally using density functional calculations in order to understand the nature of bonding between phosphorus and iron at grain boundaries. The values of binding energies of cluster are increasing with the phosphorous substitutions. The increases are due to Fe-P bond strengthened by the charge transfer from Fe atom to P atom. On the other hand the calculated bond orders give that Fe-Fe bonds are weakened. Thus, the embrittlement induced with the segregation of P due to irradiation is considered to be associated with weakened Fe-Fe bonds.
Tsuru, Tomohito; Abe, Yosuke; Suzuki, Chikashi; Nakazawa, Tetsuya; Kaji, Yoshiyuki; Tsukada, Takashi
Journal of Nuclear Materials, 417(1-3), p.1054 - 1057, 2011/10
Times Cited Count:1 Percentile:10.27(Materials Science, Multidisciplinary)Reduced activation ferritic steel is one of the leading structural material candidates for a nuclear fusion reactor. Since the solute impurities have an effect on the embrittlement through segregation under irradiation, the stability of impurity elements should be elaborated. In the present study the segregation characteristics of tungsten and some general solute impurities in bcc iron were investigated nonempirically by first principle calculations, where the equilibrium segregation was considered via a regular solution model and the change in enthalpy for segregation were directly evaluated for comparison. Subsequently the energetic stabilities of impurity-impurity and impurity-vacancy pair were evaluated. The segregation enthalpy is influenced by the electronic interaction between the d-electron of Fe and the outer electron of the impurity element, and molybdenum and tungsten tend to prevent from the impurity segregation.
 clusters (n 6) with Cr substitutions; UB3LYP/LanL2DZ calculation
clusters (n 6) with Cr substitutions; UB3LYP/LanL2DZ calculationNakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 50(3), p.982 - 990, 2011/01
Times Cited Count:10 Percentile:31.89(Materials Science, Multidisciplinary)Geometric parameters, binding energies, natural populations, natural electron configurations and magnetic moments are obtained for the clusters of Fe, Cr and FeCr ( = 2-6, = 1-6) optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The substitutional effects of Cr in Fen are found in the properties. The binding energies of Fen are generally decreased by successive substitutions of Cr for Fe atoms in the clusters. In the mixed Fe-Cr clusters most of Cr-Cr bond lengths are larger than the Fe-Fe and Fe-Cr bond lengths. Among the Fe-Fe, Fe-Cr and Cr-Cr bond lengths in the mixed clusters, the trend is found to become larger in that order. The larger distances between atoms in the mixed clusters are mostly caused by the strong repulsion due to magnetic frustration between atoms. The changes are associated with those of electronic structure by the Cr substitutions, especially with the extent of contribution of 4s and 3d electrons to bond.
Nakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 46(2), p.367 - 375, 2009/08
Times Cited Count:26 Percentile:59.21(Materials Science, Multidisciplinary)The clusters of Fe, Ni, and Fe-Ni are investigated computationally using a density functional approach. The geometries of clusters are optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The equilibrium geometries and binding energies are presented and discussed, together with natural populations and natural electron configurations. In addition, the binding energies of FeNi clusters are found to generally decrease by successive substitutions of Ni atoms for Fe atoms. For Fe Ni clusters, the comparisons on total energies between isomers indicate that Ni atoms energetically prefer clustering in the mixed Fe-Ni clusters. The calculations for FeNi clusters show that the clustering leads to a segregation of Ni atoms from Fe atoms.
Kaji, Yoshiyuki; Igarashi, Takahiro; Tsuru, Tomohito; Nakazawa, Tetsuya
Proceedings of 16th Pacific Basin Nuclear Conference (PBNC-16) (CD-ROM), 6 Pages, 2008/10
The intergranular stress corrosion cracking (IGSCC) is one of the critical concerns when stainless steel components have been in service in light water reactors for a long period. In order to investigate the mechanism of IGSCC, the IGSCC growth model has been developed to simulate IGSCC growth behavior including branching cracks. The present status of the IGSCC growth model and fundamental research activities for the characteristics of grain boundary is discussed in this paper.
TiONakazawa, Tetsuya; Naito, Akira*; Aruga, Takeo; Grismanovs, V.*; Chimi, Yasuhiro; Iwase, Akihiro*; Jitsukawa, Shiro
Journal of Nuclear Materials, 367-370(2), p.1398 - 1403, 2007/08
Times Cited Count:45 Percentile:92.66(Materials Science, Multidisciplinary)no abstracts in English
Kawamura, Hiroshi; Ishitsuka, Etsuo; Tsuchiya, Kunihiko; Nakamichi, Masaru; Uchida, Munenori*; Yamada, Hirokazu*; Nakamura, Kazuyuki; Ito, Haruhiko; Nakazawa, Tetsuya; Takahashi, Heishichiro*; et al.
Nuclear Fusion, 43(8), p.675 - 680, 2003/08
Times Cited Count:29 Percentile:64.03(Physics, Fluids & Plasmas)no abstracts in English
TiO irradiated with high energy ionsNakazawa, Tetsuya; Grismanovs, V.*; Yamaki, Daiju; Katano, Yoshio*; Aruga, Takeo
Nuclear Instruments and Methods in Physics Research B, 206, p.166 - 170, 2003/05
Times Cited Count:33 Percentile:87.41(Instruments & Instrumentation)LiTiO samples with high energy oxygen ions were examined by the use of Raman spectroscopy, X-ray diffraction (XRD) and scanning electron microscopy (SEM). The Raman spectra of the irradiated samples do not show any evidence of destruction of LiTiO structure. On the other hand, the Bragg peak intensity of XRD is decreased with the increasing the ion fluence. Especially, the intensity of (002) supercell peak is drastically reduced after the irradiation to 1.2E+19 ions/m . This result implies that partial site mixing between Li and Ti atoms is induced by the irradiation. The transition to such a disordering is observed also by SEM examination; the grain structure at surface layer is vanished after the irradiation.
. This result implies that partial site mixing between Li and Ti atoms is induced by the irradiation. The transition to such a disordering is observed also by SEM examination; the grain structure at surface layer is vanished after the irradiation.
with surface hydroxyl groups in lithium silicatesNakazawa, Tetsuya; Yokoyama, Keiichi; Grismanovs, V.*; Katano, Yoshio*; Jitsukawa, Shiro
Journal of Nuclear Materials, 307-311(Part2), p.1436 - 1440, 2002/12
Times Cited Count:1 Percentile:9.70(Materials Science, Multidisciplinary)no abstracts in English
Nakazawa, Tetsuya; Yokoyama, Keiichi; Grismanovs, V.*; Katano, Yoshio*; Jitsukawa, Shiro
Journal of Nuclear Materials, 302(2-3), p.165 - 174, 2002/04
Times Cited Count:3 Percentile:22.58(Materials Science, Multidisciplinary)no abstracts in English
O molecules from surface hydroxyl groups in silicatesNakazawa, Tetsuya; Yokoyama, Keiichi; Grismanovs, V.*; Katano, Yoshio*
Journal of Nuclear Materials, 297(1), p.69 - 76, 2001/07
Times Cited Count:9 Percentile:55.14(Materials Science, Multidisciplinary)no abstracts in English
OKatano, Yoshio*; Aruga, Takeo; Yamamoto, Shunya; Nakazawa, Tetsuya; Yamaki, Daiju; Noda, Kenji
Journal of Nuclear Materials, 283-287(Part.2), p.942 - 946, 2000/12
Times Cited Count:0 Percentile:0.00(Materials Science, Multidisciplinary)no abstracts in English
Nakazawa, Tetsuya; Yokoyama, Keiichi; Grismanous, J.*; Katano, Yoshio*
Journal of Nuclear Materials, 279(2-3), p.201 - 206, 2000/06
Times Cited Count:10 Percentile:56.42(Materials Science, Multidisciplinary)no abstracts in English
O irradiated with triple beam of H, He and heavy ionsKatano, Yoshio*; Aruga, Takeo; Yamamoto, Shunya; Nakazawa, Tetsuya; Yamaki, Daiju
Proceedings of 2000 International Conference on Ion Implantation Technology (IIT 2000), p.805 - 808, 2000/00
no abstracts in English
