


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
天野 由記; Sachdeva, R.*; Gittins, D.*; Anantharaman, K.*; Lei, S.*; Valentin-Alvarado, L. E.*; Diamond, S.*; 別部 光里*; 岩月 輝希; 望月 陽人; et al.
Environmental Microbiome (Internet), 19, p.105_1 - 105_17, 2024/12
被引用回数:0 パーセンタイル:0.00(Genetics & Heredity)Underground research laboratories (URLs) provide a window on the deep biosphere and enable investigation of potential microbial impacts on nuclear waste, CO and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO-rich Horonobe URL. We detected near-identical genotypes for approximately one third of all genomically defined organisms at multiple depths within the Horonobe URL. This cannot be explained by inactivity, as in situ growth was detected for some bacteria, albeit at slow rates. Given the current low hydraulic conductivity and groundwater compositional heterogeneity, ongoing inter-site strain dispersal seems unlikely. Alternatively, the Horonobe URL microbiome homogeneity may be explained by higher groundwater mobility during the last glacial period. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates between widely separated underground sites may be fast enough relative to mutation rates to have precluded substantial divergence in species composition. Species overlaps between subsurface locations on different continents constrain expectations regarding the scale of global subsurface biodiversity. Overall, microbiome and geochemical stability over the study period has important implications for underground storage applications.
and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO-rich Horonobe URL. We detected near-identical genotypes for approximately one third of all genomically defined organisms at multiple depths within the Horonobe URL. This cannot be explained by inactivity, as in situ growth was detected for some bacteria, albeit at slow rates. Given the current low hydraulic conductivity and groundwater compositional heterogeneity, ongoing inter-site strain dispersal seems unlikely. Alternatively, the Horonobe URL microbiome homogeneity may be explained by higher groundwater mobility during the last glacial period. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates between widely separated underground sites may be fast enough relative to mutation rates to have precluded substantial divergence in species composition. Species overlaps between subsurface locations on different continents constrain expectations regarding the scale of global subsurface biodiversity. Overall, microbiome and geochemical stability over the study period has important implications for underground storage applications.
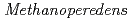 archaea host diverse and interacting extrachromosomal elements
archaea host diverse and interacting extrachromosomal elementsShi, L.-D.*; West-Roberts, J.*; Schoelmerich, M. C.*; Penev, P. I.*; Chen, L.-X.*; 天野 由記; Lei, S.*; Sachdeva, R.*; Banfield, J. F.*
Nature Microbiology (Internet), 9(9), p.2422 - 2433, 2024/09
被引用回数:1 パーセンタイル:45.91(Microbiology)Methane emissions that contribute to climate change can be mitigated by anaerobic methane-oxidizing archaea such as . Some have huge extrachromosomal genetic elements (ECEs) called Borgs that may modulate their activity, yet the broader diversity of ECEs is little studied. Here, we report small enigmatic linear ECEs, circular viruses and unclassified ECEs, that we predict replicate within . The linear ECEs have features such as inverted terminal repeats, pervasive tandem repeats, and coding patterns that are strongly reminiscent of Borgs, but they are only 52 kb to 145 kb in length. They share proteins with Borgs and . Thus, we refer to them as mini-Borgs. Mini-Borgs are genetically diverse and we assign them to at least five family-level groups. We also identify eight novel families of viruses, some of which encode multiheme cytochromes, and unclassified circular ECEs that encode TnpB genes. A population-heterogeneous CRISPR array is encoded in close proximity to TnpB and has spacers that target other ECEs including previously reported plasmids. The diverse groups of ECEs exchange genetic information with each other and with , likely impacting the activity and evolution of these environmentally important archaea.
archaeaSchoelmerich, M. C.*; Oubouter, H. T.*; Sachdeva, R.*; Penev, P. I.*; 天野 由記; West-Roberts, J.*; Welte, C. U.*; Banfield, J. F.*
Nature Communications (Internet), 13, p.7085_1 - 7085_11, 2022/11
被引用回数:12 パーセンタイル:73.73(Multidisciplinary Sciences)Anaerobic methanotrophic archaea (ANME) make energy from the breakdown of methane, an important driver of global warming, yet the extrachromosomal genetic elements that impact the activities of ANME are little understood. Here we describe large plasmids associated with ANME of the Methanoperedens Genus. These have been maintained in two enrichment bioreactors that are highly dominated by different species and associate with species in other anaerobic environments. By manual curation we show that two of the plasmids are large (155,607 bp and 191,912 bp), circular, and replicate bidirectionally. The group of species that carry these plasmids is related to  ,
,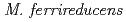 and
and 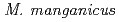 and the plasmids occur in the same copy number as the main chromosome. The larger plasmid encodes transporters that potentially enhance access to Ni, which is required for the MCR complex, Co required for the cobalamin cofactor needed for methyltransferases tied to central processes and amino acid uptake. We show that many plasmid genes are actively transcribed, including genes involved in plasmid chromosome maintenance and segregation, a Co
and the plasmids occur in the same copy number as the main chromosome. The larger plasmid encodes transporters that potentially enhance access to Ni, which is required for the MCR complex, Co required for the cobalamin cofactor needed for methyltransferases tied to central processes and amino acid uptake. We show that many plasmid genes are actively transcribed, including genes involved in plasmid chromosome maintenance and segregation, a Co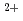 /Ni transporter and cell protective proteins. Notably, plasmid-borne genes for a ribosomal protein uL16 and adjacent elongation factor eEF2 are highly expressed. These are not encoded in the host genome, indicating an obligate interdependence between this plasmid and its host. The finding of plasmids of opens the way for the development of genetic vectors that could be used to probe little understood aspects of physiology. Ultimately, this may provide a route to introduce or alter genes that may enhance growth and overall metabolism to accelerate methane oxidation rates.
/Ni transporter and cell protective proteins. Notably, plasmid-borne genes for a ribosomal protein uL16 and adjacent elongation factor eEF2 are highly expressed. These are not encoded in the host genome, indicating an obligate interdependence between this plasmid and its host. The finding of plasmids of opens the way for the development of genetic vectors that could be used to probe little understood aspects of physiology. Ultimately, this may provide a route to introduce or alter genes that may enhance growth and overall metabolism to accelerate methane oxidation rates.
Jaffe, A. L.*; Thomas, A. D.*; He, C.*; Keren, R.*; Valentin-Alvarado, L. E.*; Munk, P.*; Bouma-Gregson, K.*; Farag, I. F.*; 天野 由記; Sachdeva, R.*; et al.
mBio, 12(4), p.e00521-21_1 - e00521-21_21, 2021/08
被引用回数:27 パーセンタイル:87.79(Microbiology)Candidate Phyla Radiation (CPR) bacteria are small, likely episymbiotic organisms found across Earth's ecosystems. Despite their prevalence, the distribution of CPR lineages across habitats and the genomic signatures of transitions amongst these habitats remain unclear. Hear, we expand the genome inventory for Absconditabacteria (SR1), Gracilibacteria, and Saccharibacteria (TM7), CPR bacteria known to occur in both animal-associated and environmental microbiomes, and investigate variation in gene content with habitat of origin. By overlaying phylogeny with habitat information, we show that bacteria from these three lineages have undergone multiple transitions from environmental habitats into animal microbiomes. Based on co-occurrence analyses of hundreds of metagenomes, we extend the prior suggestion that certain TM7 have broad bacterial host ranges and constrain possible host relationships for SR1 and Gracilibacteria. Full-proteome analyses show that animal-associated TM7 have smaller gene repertoires than their environmental counterparts and are enriched in numerous protein families, including those likely functioning in amino acid metabolism, phage defense, and detoxification of peroxide. In contrast, some freshwater TM7 encodea putative rhodopsin. For protein families exhibiting the clearest patterns of differential habitat distribution, we compared protein and species phylogenies to estimate the incidence of lateral gene transfer and genomic loss occurring over the species tree. These analyses suggest that habitat transitions were likely not accompanied by large transfer or loss events, but rather were associated with continuous proteome remodeling. Thus, we speculate that CPR habitat transitions were driven largely by availability of suitable host taxa, and were reinforced by acquisition and loss of some capacities.
Al-Shayeb, B.*; Sachdeva, R.*; Chen, L.-X.*; Ward, F.*; Munk, P.*; Devoto, A.*; Castelle, C. J.*; Olm, M. R.*; Bouma-Gregson, K.*; 天野 由記; et al.
Nature, 578(7795), p.425 - 431, 2020/02
被引用回数:291 パーセンタイル:99.46(Multidisciplinary Sciences)Phage typically have small genomes and depend on their bacterial hosts for replication. We generated metagenomic datasets from many diverse ecosystems and reconstructed hundreds of huge phage genomes, between 200 kbp and 716 kbp in length. Thirty four genomes were manually curated to completion, including the largest phage genomes yet reported. Expanded genetic repertoires include diverse and new CRISPR-Cas systems, tRNAs, tRNA synthetases, tRNA modification enzymes, initiation and elongation factors and ribosomal proteins. Phage CRISPR have the capacity to silence host transcription factors and translational genes, potentially as part of a larger interaction network that intercepts translation to redirect biosynthesis to phage-encoded functions. Some phage repurpose bacterial systems for phage-defense to eliminate competing phage. We phylogenetically define seven major clades of huge phage from human and other animal microbiomes, oceans, lakes, sediments, soils and the built environment. We conclude that large gene inventories reflect a conserved biological strategy, observed across a broad bacterial host range and resulting in the distribution of huge phage across Earth's ecosystems.
天野 由記; Sachdeva, R.*; Gittins, D.*; Anantharaman, K.*; Lei, S.*; 別部 光里*; 望月 陽人; Thomas, B. C.*; 鈴木 庸平*; Banfield, J. F.*
no journal, ,
Underground research laboratories (URLs) provide a window on the deep biosphere and enable investigation of potential microbial impacts on geological disposal, CO and H stored in the subsurface. We carried out the first multi-year study of groundwater microbiomes sampled from defined intervals between 140 and 400 m below the surface of the Horonobe and Mizunami URLs, Japan. We reconstructed draft genomes for  90% of all organisms detected over a four-year period. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO -rich Horonobe URL. High fluid flow zones and proximity to subsurface tunnels apparently impact microbial community composition. We detected near-identical genotypes for approximately one third of genomically defined organisms at multiple depths within the Horonobe URL. Sequencing data indicate that at least some of the bacteria are growing, albeit slowly. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates may be fast enough relative to mutation rates to have limited substantial species divergence. Hydraulic and isotopic measurements predict inter-site movement of groundwater over thousands of years, but it is uncertain whether the permeability is sufficient for transport of microbial cells. Alternatively, Horonobe URL microbiome homogeneity may be explained by dispersal during last glacial period.
90% of all organisms detected over a four-year period. The Horonobe and Mizunami microbiomes are dissimilar, likely because the Mizunami URL is hosted in granitic rock and the Horonobe URL in sedimentary rock. Despite this, hydrogen metabolism, rubisco-based CO fixation, reduction of nitrogen compounds and sulfate reduction are well represented functions in microbiomes from both URLs, although methane metabolism is more prevalent at the organic- and CO -rich Horonobe URL. High fluid flow zones and proximity to subsurface tunnels apparently impact microbial community composition. We detected near-identical genotypes for approximately one third of genomically defined organisms at multiple depths within the Horonobe URL. Sequencing data indicate that at least some of the bacteria are growing, albeit slowly. Genotypically-defined species closely related to those detected in the URLs were identified in three other subsurface environments in the USA. Thus, dispersal rates may be fast enough relative to mutation rates to have limited substantial species divergence. Hydraulic and isotopic measurements predict inter-site movement of groundwater over thousands of years, but it is uncertain whether the permeability is sufficient for transport of microbial cells. Alternatively, Horonobe URL microbiome homogeneity may be explained by dispersal during last glacial period.
