


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Ikebe, Kimiyoshi; Sakuraba, Shun*; Kono, Hidetoshi
PLOS Computational Biology, 12(3), p.e1004788_1 - e1004788_13, 2016/03
Times Cited Count:50 Percentile:90.68(Biochemical Research Methods)Sakuraba, Shun
Tokei Suri, 62(2), p.171 - 184, 2014/12
In this review, we explain dimensionality reduction techniques applied in the field of the biomolecular simulation. The methods based on the linear transformation are reviewed. We also investigate how these methods use correlation for reducing the dimension of biomolecules. Our review includes functional mode analysis, principal component analysis, full correlation analysis, quasi anharmonic analysis, and independent subspace analysis. We also discuss the physical interpretation and implications of the methods.
Li, H.*; Sakuraba, Shun; Chandrasekaran, A.*; Yang, L.-W.*
Journal of Chemical Information and Modeling, 54(8), p.2275 - 2285, 2014/08
Times Cited Count:17 Percentile:69.13(Chemistry, Medicinal)We provide evidence supporting that protein-protein and protein-ligand docking poses are functions of protein shape and intrinsic dynamics. Over sets of 68 protein-protein complexes and 240 non-homologous enzymes, we recognize common predispositions for binding sites to have minimal vibrations and angular momenta while two interacting proteins orient so as to maximize the angle between their rotation/bending axes (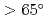 ). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into "domains" of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
). The findings are then used to define quantitative criteria to filter out docking decoys less likely to be the near-native poses, hence the chances to find near-native hits can be doubled. With the novel approach to partition a protein into "domains" of robust but disparate intrinsic dynamics, 90% of catalytic residues in enzymes can be found within the first 50% of the residues closest to the interface of these dynamics domains. The results suggest an anisotropic rather than isotropic distribution of catalytic residues near the mass centers of enzymes.
Sakuraba, Shun; Matsubayashi, Nobuyuki*
Journal of Computational Chemistry, 35(21), p.1592 - 1608, 2014/08
Times Cited Count:61 Percentile:80.66(Chemistry, Multidisciplinary)ERmod is a software package to efficiently and approximately compute the solvation free energy using the method of energy representation. Molecular simulation is to be conducted at two condensed-phase systems of the solution of interest and the reference solvent with test-particle insertion of the solute. The subprogram 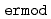 in ERmod then provides a set of energy distribution functions from the simulation trajectories, and another subprogram
in ERmod then provides a set of energy distribution functions from the simulation trajectories, and another subprogram 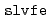 determines the solvation free energy from the distribution functions through an approximate functional. The present paper describes the design and implementation of ERmod, and illustrates its performance in solvent water for two organic solutes and two protein solutes. Actually, the free-energy computation with ERmod is not restricted to the solvation in homogeneous medium such as fluid and polymer, and can treat the binding into weakly ordered system with nano-inhomogeneity such as micelle and lipid membrane. ERmod is available on web at
determines the solvation free energy from the distribution functions through an approximate functional. The present paper describes the design and implementation of ERmod, and illustrates its performance in solvent water for two organic solutes and two protein solutes. Actually, the free-energy computation with ERmod is not restricted to the solvation in homogeneous medium such as fluid and polymer, and can treat the binding into weakly ordered system with nano-inhomogeneity such as micelle and lipid membrane. ERmod is available on web at  .
.
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
Journal of Computational Chemistry, 35(1), p.39 - 50, 2014/01
Times Cited Count:17 Percentile:44.03(Chemistry, Multidisciplinary)Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
no abstracts in English
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
no abstracts in English
Ikebe, Kimiyoshi; Sakuraba, Shun*; Kono, Hidetoshi
no journal, ,
Kono, Hidetoshi; Ikebe, Kimiyoshi; Sakuraba, Shun*; Ishida, Hisashi
no journal, ,
Kono, Hidetoshi; Ikebe, Kimiyoshi; Sakuraba, Shun*; Ishida, Hisashi
no journal, ,
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
Kono, Hidetoshi; Sakuraba, Shun; Ishida, Hisashi
no journal, ,
Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
Upon analyzing molecular dynamics simulation, two or more simulation results are often compared - it is also the case with biomolecular simulations. Comparing the effect of the mutations, pressure change or ligand binding, is the common example of such cases, to name a few. In this presentation we will show that an existing method, called Linear Discriminant Analysis (LDA), is useful for comparing several simulations. Applications and the comparison to the existing methods will be presented.
Kono, Hidetoshi; Sakuraba, Shun; Ishida, Hisashi
no journal, ,
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
no abstracts in English
Ikebe, Kimiyoshi; Sakuraba, Shun*; Kono, Hidetoshi
no journal, ,
Ikebe, Kimiyoshi; Sakuraba, Shun*; Kono, Hidetoshi
no journal, ,
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
no abstracts in English
Kono, Hidetoshi; Ikebe, Kimiyoshi; Li, Z.; Sakuraba, Shun*
no journal, ,
no abstracts in English
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
no journal, ,
