


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metals
path-integral simulations of hydrogen-isotope diffusion in face-centred cubic metalsKimizuka, Hajime*; Ogata, Shigenobu*; Thomsen, B.; Shiga, Motoyuki
Journal of Physics; Condensed Matter, 37(19), p.193001_1 - 193001_16, 2025/04
Times Cited Count:0 Percentile:0.00(Physics, Condensed Matter)Calculations of hydrogen isotope diffusion coefficients for face-centred cubic lattice metals using  path integral methods show that the temperature dependence of the diffusion coefficients has an unusual shape. This is due to the competition between nuclear quantum effects with different temperature dependence, which explains the unusual crossover mechanism between the normal and inverse isotope effects. We also present the results of a path integral simulation that describes approximate quantum dynamics using machine learning-based interatomic potentials with almost the same accuracy as density functional theory calculations. This computational method paves the way for understanding the quantum behaviour of hydrogen isotopes in various solid-state materials.
path integral methods show that the temperature dependence of the diffusion coefficients has an unusual shape. This is due to the competition between nuclear quantum effects with different temperature dependence, which explains the unusual crossover mechanism between the normal and inverse isotope effects. We also present the results of a path integral simulation that describes approximate quantum dynamics using machine learning-based interatomic potentials with almost the same accuracy as density functional theory calculations. This computational method paves the way for understanding the quantum behaviour of hydrogen isotopes in various solid-state materials.
and machine learning potentials for modeling nuclear quantum effects in waterThomsen, B.; Nagai, Yuki*; Kobayashi, Keita; Hamada, Ikutaro*; Shiga, Motoyuki
Journal of Chemical Physics, 161(20), p.204109_1 - 204109_18, 2024/11
Times Cited Count:1 Percentile:37.13(Chemistry, Physical)We introduce the self-learning path integral hybrid Monte Carlo with mixed and machine learning potentials (SL-PIHMC-MIX) method which allows the application of hybrid Monte Carlo for both path integrals and for larger system sizes. The method shows savings of an order of magnitude with respect to the number of DFT calculations needed to calculate and converge the structure of room temperature water when using SL-PIHMC-MIX over ab initio path integral molecular dynamics (PIMD).
Nakata, Yuto; Sasaki, Takehiko*; Thomsen, B.; Shiga, Motoyuki
Chemical Physics Letters, 845, p.141285_1 - 141285_9, 2024/06
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Using density functional theory and metadynamics simulations, we study cellobiose hydrolysis and glucose hydrogenation with silica-supported platinum and palladium catalysts in hot water, relevant to green cellulose conversion. It is found that cellobiose hydrolysis can proceed by the attack of hydrogen atoms adsorbed on metal or protons spilled over to silica forming glucose. Glucose can then be hydrogenated by hydrogen atoms adsorbed at platinum/water interface forming sorbitol. The reaction barriers of hydrolysis and hydrogenation at platinum/water interface are both lower than that at palladium/water interface, which explains the experimental finding that the platinum performs as a better catalyst than palladium.
Tsuchiya, Jun*; Shiga, Motoyuki; Tsuneyuki, Shinji*; Thompson, E. C.*
Physical Review Research (Internet), 6(2), p.023302_1 - 023302_6, 2024/06
We investigate the effect of nuclear quantum effects (NQEs) of hydrogen atoms on the elasticity of ice VII at high pressure and ambient temperature conditions using ab initio path-integral molecular dynamics (PIMD) calculations. We find that the NQEs of hydrogen contributes to the transition of ice VII from a static disordered structure to a dynamically disordered structure at pressures exceeding 40 GPa. This transition is marked by a discontinuous increase of the elastic constants. Comparison of molecular dynamics and PIMD calculations reveal that NQEs increase the elastic constants of ice by about 20% at 70 GPa and 300 K.
Nagai, Yuki; Iwasaki, Yutaka*; Kitahara, Koichi*; Takagiwa, Yoshiki*; Kimura, Kaoru*; Shiga, Motoyuki
Physical Review Letters, 132(19), p.196301_1 - 196301_6, 2024/05
Times Cited Count:6 Percentile:82.50(Physics, Multidisciplinary)A quasicrystal is an ordered but non-periodic structure understood as a projection from a higher dimensional periodic structure. An anomalous increase in heat capacity at high temperatures has been discussed for over two decades as a manifestation of a hidden high dimensionality of quasicrystals. A theoretical study of the heat capacity of realistic quasicrystals or their approximants has yet to be conducted because of the huge computational complexity. To bridge this gap between experiment and theory, we show experiments and cutting-edge machine-learning molecular simulations on the same material, an Al-Pd-Ru quasicrystal, and its approximants. We show that at high temperatures, aluminum atoms diffuse with discontinuous-like jumps, and the diffusion paths of the aluminum can be understood in terms of jumps corresponding to hyperatomic fluctuations in six-dimensional space.
Shiga, Motoyuki; Thomsen, B.; Kimizuka, Hajime*
Physical Review B, 109(5), p.054303_1 - 054303_12, 2024/02
Times Cited Count:3 Percentile:70.50(Materials Science, Multidisciplinary)Inelastic neutron scattering spectra of hydrogen in palladium were calculated considering nuclear quantum effects at finite temperatures. A computational method combining semiclassical molecular dynamics based on path integrals and machine learning potentials was used. The calculated spectra agree well with the experimental spectra with respect to the positions and intensities of the peaks corresponding to the fundamental and first harmonic of the vibrational excitation of hydrogen atoms. Comparison with classical molecular dynamics shows that nuclear quantum effects play an essential role in the inelastic neutron scattering spectra.
Shiga, Motoyuki; Thomsen, B.; Nagai, Yuki
Ansanburu, 25(4), p.303 - 310, 2023/10
The parallel molecular simulation software "PIMD" will be presented. The use of PIMD will be explained through specific examples such as water structure by ab initio path integral molecular dynamics, quantum diffusion of hydrogen in metal by ring polymer molecular dynamics, machine learning potential generation and phonon properties of superconductors, and polyalcohol dehydration reaction by metadynamics.
Kwon, H.*; Shiga, Motoyuki; Kimizuka, Hajime*; Oda, Takuji*
Acta Materialia, 247, p.118739_1 - 118739_11, 2023/04
Times Cited Count:23 Percentile:94.28(Materials Science, Multidisciplinary)We estimate the diffusivity of dilute hydrogen in body-centered-cubic metals, Nb, Fe, and W, from path integral simulations using machine-learning moment tensor potentials with an accuracy level of density functional theory. Our computational results show great agreement with some experimental results that appear to be accurate. The isotope effects are also reproduced consistently with the experimental data.
Shiga, Motoyuki
Journal of Computational Chemistry, 43(27), p.1864 - 1879, 2022/10
Times Cited Count:7 Percentile:47.13(Chemistry, Multidisciplinary)A new approximate method for quantum vibrational dynamics (Brownian chain molecular dynamics: BCMD) based on the path integral method is proposed. Conventional methods such as centroid molecular dynamics and ring polymer dynamics cannot correctly calculate quantum vibration spectra under low temperature conditions. Thus, I introduce an over-damped Langevin equation for the non-centroid degrees of freedom of atoms to suppress unphysical resonances and vibrational rotational couplings, which are the source of the problem. I verify this approximation through applications to infrared absorption spectra of light and heavy water. I also demonstrate the feasibility of ab initio BCMD simulations by the combination with electronic structure theory.
Kimizuka, Hajime*; Thomsen, B.; Shiga, Motoyuki
Journal of Physics; Energy (Internet), 4(3), p.034004_1 - 034004_13, 2022/07
Times Cited Count:20 Percentile:77.13(Chemistry, Physical)Artificial neural network-based interatomic potential for a system of palladium and hydrogen was developed, and path integral molecular dynamics simulations were performed to study the quantum diffusion of hydrogen isotopes in palladium crystals. Diffusion coefficients of light and heavy hydrogen were calculated over a wide temperature range of 50-1500 K to clarify the difference in diffusion mechanisms at low and high temperatures.
path integral simulationsThomsen, B.; Shiga, Motoyuki
Physical Chemistry Chemical Physics, 24(18), p.10851 - 10859, 2022/05
Times Cited Count:6 Percentile:42.36(Chemistry, Physical)Akazawa, Daisuke; Sasaki, Takehiko*; Nagasaka, Masanari*; Shiga, Motoyuki
Journal of Chemical Physics, 156(4), p.044202_1 - 044202_7, 2022/01
Times Cited Count:8 Percentile:62.23(Chemistry, Physical)The hydration structure of cellulose is very important for understanding the hydrolysis of cellulose at the molecular level. In this paper, we report a joint experimental and theoretical study on the X-ray absorption spectroscopy (XAS) of aqueous cellobiose, a disaccharide unit of cellulose. In the experimental part, high resolution measurements of the carbon K-edge XAS spectra were performed. It was found that the peak heights in the spectrum change considerably over the temperature range of 25  C to 60 C, which is a reflection of the number of hydrogen bonds between cellobiose and water. We suggest that this spectral change could be useful information for identifying the hydration of cellulose in various environments.
C to 60 C, which is a reflection of the number of hydrogen bonds between cellobiose and water. We suggest that this spectral change could be useful information for identifying the hydration of cellulose in various environments.
study of nuclear quantum effects on sub- and supercritical waterThomsen, B.; Shiga, Motoyuki
Journal of Chemical Physics, 155(19), p.194107_1 - 194107_11, 2021/11
Times Cited Count:8 Percentile:47.53(Chemistry, Physical)Pal, A.*; Pal, S.*; Verma, S.*; Shiga, Motoyuki; Nair, N. N.*
Journal of Computational Chemistry, 42(28), p.1996 - 2003, 2021/10
Times Cited Count:11 Percentile:51.41(Chemistry, Multidisciplinary)Temperature-accelerated sliced sampling is a computational method for high-dimensional free energy landscapes. In the conventional method, post-processing of the calculation has been required using the weighted histogram analysis method. In this study, we establish a refined approach without post-processing using the average force. We demonstrated the cases of the two-dimensional and four-dimensional free energy landscapes of alanine dipeptide and tripeptide, where they were computed within an error of about kcal/mol. Vast application of this method is expected for free energy calculations in computational chemistry.
Kondo, Tomomi; Sasaki, Takehiko*; Shiga, Motoyuki
Journal of Computational Chemistry, 42(25), p.1783 - 1791, 2021/09
Times Cited Count:5 Percentile:24.90(Chemistry, Multidisciplinary)Sugar alcohol dehydration in hot water is an important reaction that allows for environmentally friendly biomass conversion without the use of organic solvents. Here, we report a free-energy analysis by metadynamics (MTD) simulations based on ab initio and semiempirical electronic structure theory to understand the mechanism of dehydration reactions of sorbitol (SBT) in hot acidic water. It was found that the reaction proceeds via an S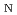 2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected by the acidic species. The free energy barriers of the reaction pathways leading to five-membered ether products are lower than those leading to six-membered ether products.
2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected by the acidic species. The free energy barriers of the reaction pathways leading to five-membered ether products are lower than those leading to six-membered ether products.
Kobayashi, Keita; Nagai, Yuki; Itakura, Mitsuhiro; Shiga, Motoyuki
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
Times Cited Count:13 Percentile:67.66(Chemistry, Physical)no abstracts in English
path integral simulationsKimizuka, Hajime*; Shiga, Motoyuki
Physical Review Materials (Internet), 5(6), p.065406_1 - 065406_9, 2021/06
Times Cited Count:15 Percentile:57.57(Materials Science, Multidisciplinary)Nuclear quantum effects are a non-negligible factor in the dynamic behavior of hydrogen in metals. In this study, we investigated the hydrogen diffusion in the face-centered cubic metals Al, Ag, and Cu using a first-principles integral molecular dynamics simulation that takes into account the nuclear quantum effects. It was found that the temperature dependence of hydrogen diffusion in Ag and Cu is inverted S-shaped, while the temperature dependence of hydrogen diffusion in Al is C-shaped. This difference is due to the fact that the most stable position of hydrogen is the octahedral site in Ag and Cu, while it is the tetrahedral site in Al. Therefore, it is found that the nuclear quantum effects of hydrogen diffusion (zero-point oscillation and tunneling) differ qualitatively depending on metals with different stable sites.
path integral molecular dynamicsThomsen, B.; Shiga, Motoyuki
Journal of Chemical Physics, 154(8), p.084117_1 - 084117_10, 2021/02
Times Cited Count:14 Percentile:70.46(Chemistry, Physical)In this study we investigate the nuclear quantum effects on the acidity constant of liquid water isotopologues at the ambient condition by path integral molecular dynamics simulations. This technique not only reproduces the acidity constants of liquid D O experimentally measured but also allows for a theoretical prediction of the acidity constants of liquid TO, aqueous HDO and HTO, which are unknown due to its scarcity. The results indicate that the nuclear quantum effects play an indispensable role in the absolute determination of acidity constants.
O experimentally measured but also allows for a theoretical prediction of the acidity constants of liquid TO, aqueous HDO and HTO, which are unknown due to its scarcity. The results indicate that the nuclear quantum effects play an indispensable role in the absolute determination of acidity constants.
Kondo, Tomomi; Sasaki, Takehiko*; Ruiz-Barragan, S.*; Ribas-Ari o, J.*; Shiga, Motoyuki; Ruiz-Barragan, S.*
o, J.*; Shiga, Motoyuki; Ruiz-Barragan, S.*
Journal of Computational Chemistry, 42(3), p.156 - 165, 2021/01
Times Cited Count:5 Percentile:19.73(Chemistry, Multidisciplinary)We propose a canonical sampling method to refine metadynamics simulations a posteriori. This approach could be useful particularly when two or more free energy barriers are to be compared among chemical reactions in different or competing conditions. The method was then applied to study the acid dependence of polyalcohol dehydration reactions in high-temperature aqueous solutions. It was found that the reaction proceeds consistently via an S2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected significantly by the acidic species.
Noguchi, Yoshifumi*; Hiyama, Miyabi*; Shiga, Motoyuki; Akiyama, Hidefumi*; Sugino, Osamu*
Journal of Chemical Physics, 153(20), p.201103_1 - 201103_6, 2020/11
Times Cited Count:2 Percentile:8.04(Chemistry, Physical)Stabilizing mechanisms of three isomers of the aqueous oxyluciferin in the first excited state were investigated using first-principles molecular dynamics simulations. Only the phenolate-keto isomer became attracted to the water molecules in its excited state and was stabilized by increasing the number of hydrogen bonds with nearby water molecules. The most stable isomer in the excited state was the phenolate-keto, and the phenolate-enol and phenol-enolate isomers were higher in energy by 0.38 eV and 0.57 eV, respectively, than the phenolate-keto. This was in contrast to the case of ground state in which the phenolate-enol was the most stable isomer.
