


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
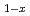 Ru
Ru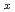 )
) Sb
Sb investigated by inelastic X-ray scattering
investigated by inelastic X-ray scatteringTsutsui, Satoshi; Kaneko, Koji; Miyazaki, Ryoichi*; Higashinaka, Ryuji*; Aoki, Yuji*; Kobayashi, Riki*; Wakimoto, Shuichi; Baron, A. Q. R.*; Sugawara, Hitoshi*; Sato, Hideyuki*
JPS Conference Proceedings (Internet), 3, p.011060_1 - 011060_5, 2014/06
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics, 394(1), p.46 - 51, 2012/02
Times Cited Count:18 Percentile:53.54(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed for porphycene and its isotopic variants in order to understand the effect of isotopic substitution of inner protons on the double proton transfer mechanism. Our quantum simulations show that double proton transfer of the unsubstituted porphycene at the room temperature mainly occurs via a so-called concerted mechanism through the second-order saddle point. In addition, we found that both isotopic substitution and temperature significantly affect the double proton transfer mechanism. For example, the contribution of the stepwise mechanism increases with a temperature increase. It has been found that out-of-plane vibrational motions significantly decrease the contribution of the concerted proton transfer mechanism.
Sugawara, Shuichi*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Physical Chemistry A, 115(42), p.11486 - 11494, 2011/09
Times Cited Count:20 Percentile:56.15(Chemistry, Physical)We have carried out path integral molecular dynamics simulations for hydrated sulfuric acid clusters to understand acid-dissociation and hydrogen-bonded structural rearrangement processes in these clusters from a quantum mechanical viewpoint. We have found that the acid dissociation processes, first and second deprotonation, effectively occur in a hydrated cluster with a specific cluster size. The mechanisms of the proton-transfer processes were analyzed in detail and it was found that the distance between O in sulfuric acid and O in the proton-accepting water is playing an important role. We also found that the water coordination number of the proton-accepting water is important in the proton-transfer processes.
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics Letters, 496(1-3), p.14 - 19, 2010/08
Times Cited Count:28 Percentile:67.01(Chemistry, Physical)Full-dimensional path-integral molecular dynamics simulations were performed to determine whether the double proton transfer tautomerization of porphycene is a concerted or a stepwise process. It was found that double proton transfer occurs dominantly through the concerted pathway via the second-order saddle point structure and that contribution of the stepwise mechanism increases with a temperature increase. Nuclear quantum effects play essential roles in determining the proton transfer mechanism.
