


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Suzuki, Chikashi; Nakajima, Kunihisa; Osaka, Masahiko
Journal of Nuclear Science and Technology, 59(3), p.345 - 356, 2022/03
Times Cited Count:3 Percentile:30.71(Nuclear Science & Technology)During a severe accident (SA) such as the Fukushima Daiichi Nuclear Power Plant accident, fission products (FP) can be retained on the surface of structural materials in reactors. Cesium (Cs) is an important FP, and various Cs compounds such as Cs silicates are formed on the surface of stainless steel (SS) in a reactor during a SA. We calculated total energies of Cs-Si-O compounds for evaluation on phase stability within an adiabatic approximation. The calculations indicate that Cs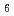 Si
Si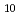 O
O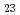 is the most stable of the Cs-Si-O compounds. We calculated, furthermore, total energies of Cs-Si-Fe-O compounds. These calculations indicate that Cs-Si-Fe-O compounds are more stable than C-Si-O compounds and that CsSi
is the most stable of the Cs-Si-O compounds. We calculated, furthermore, total energies of Cs-Si-Fe-O compounds. These calculations indicate that Cs-Si-Fe-O compounds are more stable than C-Si-O compounds and that CsSi FeO is the most stable of these C-Si-O and Cs-Si-Fe-O compounds within an adiabatic approximation. The results of our present calculations and our previous experiments lead to the conclusion that Cs-Si-Fe-O compounds can be stably formed on SS surface by Cs chemisorption.
FeO is the most stable of these C-Si-O and Cs-Si-Fe-O compounds within an adiabatic approximation. The results of our present calculations and our previous experiments lead to the conclusion that Cs-Si-Fe-O compounds can be stably formed on SS surface by Cs chemisorption.
Miwa, Shuhei; Nakajima, Kunihisa; Suzuki, Chikashi; Rizaal, M.; Suzuki, Eriko; Horiguchi, Naoki; Osaka, Masahiko
Proceedings of Joint International Conference on Supercomputing in Nuclear Applications + Monte Carlo 2020 (SNA + MC 2020), p.253 - 260, 2020/12
We have proceeded the fundamental study for the improvement of the evaluation of fission product (FP) chemistry and the treatment of fine space resolution which are main issues for the evaluation of FP behaviors in a severe accident (SA). We have been developing FP chemistry database named ECUME for the improvement of SA analysis codes. We prepared thermodynamic data for Cs compounds which is no experimental data available by computational approach. Regarding the space resolution issue, we have been developing analysis tool named CHASER based on 3D-CFD code with the model for FP chemistry. More accurate evaluation of FP behavior can be achieved by incorporating ECUME to the CHASER.
Suzuki, Chikashi; Osaka, Masahiko; Nakagiri, Toshio
Proceedings of Joint International Conference on Supercomputing in Nuclear Applications + Monte Carlo 2020 (SNA + MC 2020), p.131 - 136, 2020/10
In order to evaluate uranium migration behavior in Ningyo-toge area, we are examining the stability of uranium minerals using DFT calculation. As the first step, we investigated the crystal structure and unit cell composition of ningyoite with its structural details unclear, which is a major uranium mineral there. We analyzed the reported XRD pattern of ningyoite to evaluate a crystal structure of ningyoite using XRD analysis code. This XRD analysis indicates that the distance between U atoms is 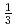
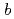 along axis. We constructed the structure of [CaU(PO
along axis. We constructed the structure of [CaU(PO )]
)] with three U or Ca atoms aligned along axis on the basis of this examination, and conducted structural optimization with this structure as initial one using DFT calculation. The theoretical XRD pattern has the maximum peak near 30
with three U or Ca atoms aligned along axis on the basis of this examination, and conducted structural optimization with this structure as initial one using DFT calculation. The theoretical XRD pattern has the maximum peak near 30 , which the reported XRD pattern of ningyoite has. This result suggests that the structure of ningyoite is based on that of [CaU(PO)].
, which the reported XRD pattern of ningyoite has. This result suggests that the structure of ningyoite is based on that of [CaU(PO)].
Miradji, F.; Suzuki, Chikashi; Nakajima, Kunihisa; Osaka, Masahiko
Journal of Physics and Chemistry of Solids, 136, p.109168_1 - 109168_9, 2020/01
Times Cited Count:4 Percentile:19.92(Chemistry, Multidisciplinary)Suzuki, Chikashi; Yaita, Tsuyoshi; Suzuki, Shinichi; Pacold, J.*; Altman, A. B.*; Minasian, S. G.*; Tyliszczak, T.*; Shuk, D. K.*; Yoshida, Hiroyuki; Osaka, Masahiko
Journal of Physics and Chemistry of Solids, 127, p.169 - 177, 2019/04
Times Cited Count:3 Percentile:13.18(Chemistry, Multidisciplinary)A combined method of NEXAFS measurement and DFT-calculation was employed for Cs evaluation in clay minerals. The Cs M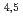 NEXAFS spectra of Cs halides were analyzed using the DFT-calculations, and were well reproduced by incorporating the core-hole strength. The Cs M NEXAFS spectrum of clay minerals was well-reproduced by the DFT-calculations including the major transitions and tail structures with the established method. The further evaluation of this spectrum by charge density suggests that these major transitions and the tail structures likely reflect bonding states and local environments around the Cs atoms. Comparison of electronic states of Cs in the clay mineral with those in the Cs halides by DFT-calculations has shown that the interaction between Cs and the nearest-neighbor atom is largest in the clay mineral, because the energy level of Cs-5s and 5p is closer to that of O-2s and 2p than the s and p orbitals of other alkali metal and alkali earth metal elements.
NEXAFS spectra of Cs halides were analyzed using the DFT-calculations, and were well reproduced by incorporating the core-hole strength. The Cs M NEXAFS spectrum of clay minerals was well-reproduced by the DFT-calculations including the major transitions and tail structures with the established method. The further evaluation of this spectrum by charge density suggests that these major transitions and the tail structures likely reflect bonding states and local environments around the Cs atoms. Comparison of electronic states of Cs in the clay mineral with those in the Cs halides by DFT-calculations has shown that the interaction between Cs and the nearest-neighbor atom is largest in the clay mineral, because the energy level of Cs-5s and 5p is closer to that of O-2s and 2p than the s and p orbitals of other alkali metal and alkali earth metal elements.
Miradji, F.; Suzuki, Chikashi; Nishioka, Shunichiro; Suzuki, Eriko; Nakajima, Kunihisa; Osaka, Masahiko; Barrachin, M.*; Do, T. M. D.*; Murakami, Kenta*; Suzuki, Masahide*
Proceedings of 9th Conference on Severe Accident Research (ERMSAR 2019) (Internet), 21 Pages, 2019/03
Osaka, Masahiko; Nakajima, Kunihisa; Miwa, Shuhei; Di Lemma, F. G.*; Miyahara, Naoya; Suzuki, Chikashi; Suzuki, Eriko; Okane, Tetsuo; Kobata, Masaaki
Proceedings of 8th European Review Meeting on Severe Accident Research (ERMSAR 2017) (Internet), 11 Pages, 2017/05
Fundamental research on fission product (FP) chemistry is underway at Japan Atomic Energy Agency. The purpose is to establish a FP chemistry database in each region of a LWR under severe accident conditions. Improvement of FP chemical models based on this database is also an important task of the research. Research outputs are reflected to the research and development of decommissioning of Fukushima Daiichi Nuclear Power Station (1F) and the enhancement of LWR safety. Four research items have thus been established considering the specific issues of 1F and the priority in the source term research area, as follows: - Effects of boron (B) release kinetics and thermal-hydraulic conditions on FP behavior, - Cesium (Cs) chemisorption and reactions with structural materials, - Establishment of a thermodynamic and thermophysical properties database for FP compounds, - Development of experimental and analytical techniques for the reproduction of FP behavior. In this paper, results and progress of the research are presented.
Osaka, Masahiko; Miwa, Shuhei; Nakajima, Kunihisa; Di Lemma, F. G.*; Suzuki, Chikashi; Miyahara, Naoya; Kobata, Masaaki; Okane, Tetsuo; Suzuki, Eriko
JAEA-Review 2016-026, 32 Pages, 2016/12
 JAEA-Review-2016-026.pdf:6.18MB
JAEA-Review-2016-026.pdf:6.18MB
A fundamental research program on fission product (FP) chemistry has started since 2012 for the purpose of establishment of a FP chemistry database in each region of LWR under severe accident and improvement of FP chemical models based on the database. Research outputs are reflected as fundamental knowledge to both the research and development of decommissioning of Fukushima Daiichi Nuclear Power Station (1F) and enhancement of LWR safety. Four research items have thus been established considering the specific issues of 1F and the priority in the source term research area, as follows: effects of boron (B) release kinetics and thermal-hydraulic conditions on FP behavior, cesium (Cs) chemisorption and reactions with structural materials, enlargement of a thermodynamic and thermophysical properties database for FP compounds and development of experimental and analytical techniques for the reproduction of FP behavior and for direct measurement methods of chemical form of FP compounds. In this report, the research results and progress for the year 2015 are described. The main accomplishment was the installation of a reproductive test facility for FP release and transport behavior. Moreover, basic knowledge about the Cs chemisorption behavior was also obtained. In addition to the four research items, a further research item is being considered for deeper interpretation of FP behavior by the analysis of samples outside of the 1F units.
Suzuki, Chikashi; Yoshida, Hiroyuki; Shuk, D. K.*; Suzuki, Shinichi; Yaita, Tsuyoshi
Proceedings of 23rd International Conference on Nuclear Engineering (ICONE-23) (DVD-ROM), 6 Pages, 2015/05
We evaluated Cs M near edge X-ray absorption fine structure (NEXAFS) of Cs halides and clay minerals containing Cs using DFT calculation with the NEXAFS of the clay minerals measured. The evaluation of Cs halides indicated that the core-hole strength (CHS) in the NEXAFS measurement was reduced to 0.6, taking into account the strength interaction of the core-hole with the valence-band electrons. On the basis of this result, Cs M NEXAFS of the clay mineral was well reproduced including the dominant peak and tail structures using DFT calculation. Moreover the calculation showed that the NEXAFS clearly changed with the displacement of Si atoms to Al in the six-membered ring of clay minerals and little did with the coexist of K atoms with Cs between layers of the clay mineral. In addition, we examined the electronic state of CsCl and the clay mineral containing Cs.
using resonant X-ray emission spectroscopy at the Ba-L and Ti K absorption edgesYoshii, Kenji; Yoneda, Yasuhiro; Jarrige, I.*; Fukuda, Tatsuo; Nishihata, Yasuo; Suzuki, Chikashi; Ito, Yoshiaki*; Terashima, Takahito*; Yoshikado, Shinzo*; Fukushima, Sei*
Journal of Physics and Chemistry of Solids, 75(3), p.339 - 343, 2014/03
Times Cited Count:10 Percentile:39.58(Chemistry, Multidisciplinary)We have studied the electronic properties of ferroelectric BaTiO using two complementary bulk-sensitive spectroscopic probes, resonant X-ray emission spectroscopy (RXES) and X-ray absorption spectroscopy in the partial fluorescence mode (PFY-XAS). Contrary to a previous study, we found no temperature change of a PFY-XAS spectrum at the Ba L edge between room temperature and 150C. This result is not supportive of the possible presence of the displacement around Ba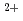 at the Curie temperature. RXES spectra were measured at the Ti K edge for BaTiO, along with SrTiO and La-doped metallic SrTiO. The photon energy of the emission peak is found to be nearly constant throughout the absorption edge for all three compounds. We deduce the Ti 3d states to have a delocalized character, in contrast with the Ba 5d states, a property which is consistent with the proposed scenario of the formation of electric dipoles in BaTiO.
at the Curie temperature. RXES spectra were measured at the Ti K edge for BaTiO, along with SrTiO and La-doped metallic SrTiO. The photon energy of the emission peak is found to be nearly constant throughout the absorption edge for all three compounds. We deduce the Ti 3d states to have a delocalized character, in contrast with the Ba 5d states, a property which is consistent with the proposed scenario of the formation of electric dipoles in BaTiO.
Suzuki, Chikashi; Nishi, Tsuyoshi; Nakada, Masami; Tsuru, Tomohito; Akabori, Mitsuo; Hirata, Masaru; Kaji, Yoshiyuki
Journal of Physics and Chemistry of Solids, 74(12), p.1769 - 1774, 2013/12
Times Cited Count:14 Percentile:51.30(Chemistry, Multidisciplinary)We investigated the electronic state of a CaF-type (Am,U) mixed oxide using the all-electron full potential linear augmented plane wave method and compared it with those of AmO, AmO, UO, and La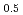 UO. The valence of Am in the mixed oxide was close to that of AmO and the valence of U in the mixed oxide was pentavalent. The electronic structure of AmO was different from that of AmO, particularly just above the Fermi level. In addition, the electronic states of Am and U in the mixed oxide were similar to those of trivalent Am and pentavalent U oxides. These electronic states reflected the high oxygen potential of AmO and the heightened oxygen potential resulting from the addition of Am to UO and also suggested the occurrence of charge transfer from Am to U in the solid solution process.
UO. The valence of Am in the mixed oxide was close to that of AmO and the valence of U in the mixed oxide was pentavalent. The electronic structure of AmO was different from that of AmO, particularly just above the Fermi level. In addition, the electronic states of Am and U in the mixed oxide were similar to those of trivalent Am and pentavalent U oxides. These electronic states reflected the high oxygen potential of AmO and the heightened oxygen potential resulting from the addition of Am to UO and also suggested the occurrence of charge transfer from Am to U in the solid solution process.
Yoneda, Yasuhiro; Yoshii, Kenji; Suzuki, Chikashi
Transactions of the Materials Research Society of Japan, 37(4), p.579 - 582, 2012/12
We performed X-ray Emission spectroscopy (XES) by using a double crystal spectrometer. Ta-L and L
and L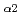 emission lines of Ta-related materials of Ta-metal, Ta
emission lines of Ta-related materials of Ta-metal, Ta O
O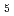 , LiTaO
, LiTaO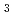 , and KTaO were measured. XES is one of the most important spectroscopies to make a remarkable progress utilizing third generation synchrotron radiation sources. Here, I discuss some aspects of XES in Ta compounds of recent experimental data. We also discuss an azimuth dependence of resonant X-ray emission spectra (RXES) of a single-crystal KTaO. The precise measurement by the double crystal spectrometer has observed change of the electronic structure by crystal orientation. Since electronic structure observation is a local probe, it can detect minute lattice deformation.
, and KTaO were measured. XES is one of the most important spectroscopies to make a remarkable progress utilizing third generation synchrotron radiation sources. Here, I discuss some aspects of XES in Ta compounds of recent experimental data. We also discuss an azimuth dependence of resonant X-ray emission spectra (RXES) of a single-crystal KTaO. The precise measurement by the double crystal spectrometer has observed change of the electronic structure by crystal orientation. Since electronic structure observation is a local probe, it can detect minute lattice deformation.
and BaSO; A Resonant X-ray emission study at the Ba-L edgeYoshii, Kenji; Jarrige, I.; Suzuki, Chikashi; Matsumura, Daiju; Nishihata, Yasuo; Yoneda, Yasuhiro; Fukuda, Tatsuo; Tamura, Kazuhisa; Ito, Yoshiaki*; Mukoyama, Takeshi*; et al.
Journal of Physics and Chemistry of Solids, 73(9), p.1106 - 1110, 2012/09
Times Cited Count:12 Percentile:44.59(Chemistry, Multidisciplinary)We have directly probed the Ba 5d states in the ferroelectric barium titanate BaTiO using resonant X-ray emission spectroscopy (RXES) and X-ray absorption spectroscopy in the partial fluorescence mode (PFY-XAS) at the Ba-L edge. The results are compared with those of the non-ferroelectric BaSO. While the RXES spectra point to a localized character for the Ba 5d states in both compounds, the main peak of the PFY-XAS spectrum, corresponding to the dipolar transitions from 2p to 5d, is found to be significantly broader for BaTiO than for BaSO. On the basis of band structure calculations, this broadening is ascribed to strong hybridization between the unoccupied Ba 5d and O 2p states in the ferroelectric. This suggests that the hybridization between the conduction states of the Ba and O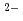 ions, and not only Ti
ions, and not only Ti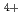 and O, plays a central role in determining the electronic structure of BaTiO.
and O, plays a central role in determining the electronic structure of BaTiO.
Suzuki, Chikashi; Nishi, Tsuyoshi; Nakada, Masami; Akabori, Mitsuo; Hirata, Masaru; Kaji, Yoshiyuki
Journal of Physics and Chemistry of Solids, 73(2), p.209 - 216, 2012/02
Times Cited Count:28 Percentile:68.56(Chemistry, Multidisciplinary)The authors investigated theoretically core-hole effects on X-ray absorption near-edge structures (XANES) of Np and Am L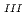 in neptunium dioxide (NpO) and americium dioxide (AmO) with CaF-type crystal lattices using the all-electron full-potential linearized augmented plane-wave (FP-LAPW) method. The peak creation mechanism of XANES was shown by examining the electronic structures of these oxides, which indicated that core-hole screening was more marked for AmO than for NpO because of the difference in the charge transfer between these oxides. Furthermore, the results of charge density analysis suggested that the white line was assigned to the quasi-bound state composed of the localized Np d or Am d components and O components, and that the tail structure was created as a result of delocalized standing waves between the Np or Am atoms.
in neptunium dioxide (NpO) and americium dioxide (AmO) with CaF-type crystal lattices using the all-electron full-potential linearized augmented plane-wave (FP-LAPW) method. The peak creation mechanism of XANES was shown by examining the electronic structures of these oxides, which indicated that core-hole screening was more marked for AmO than for NpO because of the difference in the charge transfer between these oxides. Furthermore, the results of charge density analysis suggested that the white line was assigned to the quasi-bound state composed of the localized Np d or Am d components and O components, and that the tail structure was created as a result of delocalized standing waves between the Np or Am atoms.
Igarashi, Takahiro; Nakazawa, Tetsuya; Suzuki, Chikashi; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 50(12), p.3346 - 3349, 2011/12
Times Cited Count:1 Percentile:4.27(Materials Science, Multidisciplinary)The authors have developed a new approach for large-scale systems including over 100,000 atoms to obtain physical strength from the viewpoint of atom-atom bonding energy. In this study, the authors applied this method to SiH molecule and crystalline silica system, and carried out bond order and bonding energy analyses. In this analysis, the developed method offered almost the same analytical accuracy as the first principle method, while its calculation speed was much faster than that of the latter.
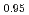 Am
Am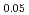 )O
)O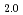
Nishi, Tsuyoshi; Nakada, Masami; Suzuki, Chikashi; Shibata, Hiroki; Okamoto, Yoshihiro; Akabori, Mitsuo; Hirata, Masaru
Journal of Nuclear Materials, 418(1-3), p.311 - 312, 2011/11
Times Cited Count:20 Percentile:79.65(Materials Science, Multidisciplinary)The XAFS measurements at U-L and Am-L absorption edge of (UAm)O were performed in transmission mode. Moreover, to clarify the valence state of Am in (U,Am)O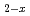 , the XANES spectrum of Am-L absorption edge of (UAm)O was verified using those of Am-L absorption edge of AmO and AmO. It was found that the XANES spectrum of the Am-L edge of (UAm)O is in good accordance with that of AmO. Thus, Am in (UAm)O is almost trivalent state.
, the XANES spectrum of Am-L absorption edge of (UAm)O was verified using those of Am-L absorption edge of AmO and AmO. It was found that the XANES spectrum of the Am-L edge of (UAm)O is in good accordance with that of AmO. Thus, Am in (UAm)O is almost trivalent state.
Tsuru, Tomohito; Abe, Yosuke; Suzuki, Chikashi; Nakazawa, Tetsuya; Kaji, Yoshiyuki; Tsukada, Takashi
Journal of Nuclear Materials, 417(1-3), p.1054 - 1057, 2011/10
Times Cited Count:1 Percentile:10.27(Materials Science, Multidisciplinary)Reduced activation ferritic steel is one of the leading structural material candidates for a nuclear fusion reactor. Since the solute impurities have an effect on the embrittlement through segregation under irradiation, the stability of impurity elements should be elaborated. In the present study the segregation characteristics of tungsten and some general solute impurities in bcc iron were investigated nonempirically by first principle calculations, where the equilibrium segregation was considered via a regular solution model and the change in enthalpy for segregation were directly evaluated for comparison. Subsequently the energetic stabilities of impurity-impurity and impurity-vacancy pair were evaluated. The segregation enthalpy is influenced by the electronic interaction between the d-electron of Fe and the outer electron of the impurity element, and molybdenum and tungsten tend to prevent from the impurity segregation.
 -Fe
-FeSuzuki, Chikashi; Tsuru, Tomohito; Kaji, Yoshiyuki
Progress in Nuclear Science and Technology (Internet), 2, p.34 - 37, 2011/10
no abstracts in English
edge in BaTiOYoshii, Kenji; Jarrige, I.; Matsumura, Daiju; Nishihata, Yasuo; Suzuki, Chikashi; Ito, Yoshiaki*; Mukoyama, Takeshi*; Tochio, Tatsunori*; Shinotsuka, Hiroshi*; Fukushima, Sei*
Japanese Journal of Applied Physics, 50(9), p.09NE03_1 - 09NE03_4, 2011/09
Times Cited Count:2 Percentile:9.27(Physics, Applied)Resonant X-ray inelastic scattering experiments have been performed at the Ba-L edge in ferroelectric BaTiO using synchrotron radiation at the BL15XU of SPring-8. An X-ray absorption spectrum of this material was found to be broader than non-ferroelectric BaSO. Molecular orbital calculations showed the formation of Ba-O bands in BaTiO, suggesting an important role of the Ba-O hybridization in the ferroelectric transition of this material. X-ray emission spectra at the edge showed a localized character of Ba-5d orbitals in BaTiO in spite of the hybridization with O-3p. A few possible applications of the measurement will be also discussed.
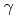 -uranium alloys; The Roll of d-d orbital interactions
-uranium alloys; The Roll of d-d orbital interactionsKurihara, Masayoshi*; Onoe, Jun*; Hirata, Masaru; Suzuki, Chikashi
Journal of Alloys and Compounds, 509(4), p.1152 - 1156, 2011/01
Times Cited Count:3 Percentile:23.28(Chemistry, Physical)The alloying behavior of transition metals (TMs) in solid -phase uranium (-U), which is expected to be used as fuel for next-generation nuclear reactors, is investigated using the discrete-variational Dirac-Fock-Slater molecular orbital method. Using a model cluster, U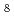 /TM, as the minimum unit of -U/TM alloys, we evaluate the d-orbital energy of the TM (Md), the bond order between the TM and U atoms, and the orbital overlap population (OOP) between the atomic orbitals of the TM and U atoms. We subsequently examine the effect of these quantities on the maximum solid solubility (MSS) of the -U/TM alloys. Md and the OOP (U 6d-TM d) exhibit good correlations with the MSS for -U/TM alloys, so that the interaction between the U-6d and TM-d atomic orbitals is found to play a key role in determining the MSS of the -U/TM alloys. The magnitude of the MSS can be explained in terms of the stabilization energy caused by U 6d-TM d orbital interactions for -U/TM alloys, which is affected by the Md and the OOP. The exponential dependence of the MSS on Md and the OOP is explained in terms of the equilibrium constant obtained using a substitution cluster model for -U/TM alloys. We also map the MSS of -U/TM alloys using the Md and the OOP as the alloying parameters. These results will assist the quantum design of nuclear fuel materials for other alloy systems besides the present alloy system.
/TM, as the minimum unit of -U/TM alloys, we evaluate the d-orbital energy of the TM (Md), the bond order between the TM and U atoms, and the orbital overlap population (OOP) between the atomic orbitals of the TM and U atoms. We subsequently examine the effect of these quantities on the maximum solid solubility (MSS) of the -U/TM alloys. Md and the OOP (U 6d-TM d) exhibit good correlations with the MSS for -U/TM alloys, so that the interaction between the U-6d and TM-d atomic orbitals is found to play a key role in determining the MSS of the -U/TM alloys. The magnitude of the MSS can be explained in terms of the stabilization energy caused by U 6d-TM d orbital interactions for -U/TM alloys, which is affected by the Md and the OOP. The exponential dependence of the MSS on Md and the OOP is explained in terms of the equilibrium constant obtained using a substitution cluster model for -U/TM alloys. We also map the MSS of -U/TM alloys using the Md and the OOP as the alloying parameters. These results will assist the quantum design of nuclear fuel materials for other alloy systems besides the present alloy system.
