


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
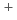 and Ag clusters
and Ag clusters小泉 亮人*; 立川 仁典*; 志賀 基之
Chemical Physics, 419, p.44 - 49, 2013/06
被引用回数:1 パーセンタイル:2.56(Chemistry, Physical)水和金属イオンに対して原子核の量子効果及び温度効果を明らかにするため、第一原理分子動力学法,経路積分分子動力学法及びリングポリマー分子動力学法を用いてAg(H O)
O)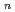 (
(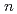 =1-4)とCu(HO)の分子構造と赤外スペクトルを調べた。その結果、量子効果及び温度効果による分子振動の非調和性は分子系や振動モードに依存していることが見いだされた。低振動モードは、複雑なモードカップリングの存在により、振動スペクトルは水和分子が多くなるにつれ幅広いバンドとなる。これに対してHOH変角振動モードのピークは、非調和性により系統的に赤方にシフトする。OH伸縮振動モードでも同様な赤方シフトが見られたが、シフトの大きさは系により依存する。したがって、上記のようなスペクトルの非調和性は、調和振動の一様なスケーリング則で表すことはできないことが結論づけられる。
=1-4)とCu(HO)の分子構造と赤外スペクトルを調べた。その結果、量子効果及び温度効果による分子振動の非調和性は分子系や振動モードに依存していることが見いだされた。低振動モードは、複雑なモードカップリングの存在により、振動スペクトルは水和分子が多くなるにつれ幅広いバンドとなる。これに対してHOH変角振動モードのピークは、非調和性により系統的に赤方にシフトする。OH伸縮振動モードでも同様な赤方シフトが見られたが、シフトの大きさは系により依存する。したがって、上記のようなスペクトルの非調和性は、調和振動の一様なスケーリング則で表すことはできないことが結論づけられる。
鈴木 机倫*; 立川 仁典*; 志賀 基之
Journal of Chemical Physics, 138(18), p.184307_1 - 184307_7, 2013/05
被引用回数:10 パーセンタイル:35.72(Chemistry, Physical)ZundelイオンH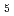 O
O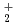 は、酸性水溶液における基本的かつ重要な分子単位の一つである。本研究では、HOとその重水素置換同位体DO及びTOの分子構造ゆらぎの量子効果と温度効果について、経路積分分子動力学シミュレーションを用いて詳しく解析した。温度100Kから900Kの範囲で調べたところ、温度上昇につれ水素結合に関係する酸素間距離,酸素水素間距離のゆらぎが急激に大きくなることを見いだした。また、結合していない水素の位置のゆらぎも温度上昇に伴い大きくなる。この温度変化はHOよりもDOやTOで大きくなるが、これは零点振動の大きさの違いに由来するものであると考えられる。
は、酸性水溶液における基本的かつ重要な分子単位の一つである。本研究では、HOとその重水素置換同位体DO及びTOの分子構造ゆらぎの量子効果と温度効果について、経路積分分子動力学シミュレーションを用いて詳しく解析した。温度100Kから900Kの範囲で調べたところ、温度上昇につれ水素結合に関係する酸素間距離,酸素水素間距離のゆらぎが急激に大きくなることを見いだした。また、結合していない水素の位置のゆらぎも温度上昇に伴い大きくなる。この温度変化はHOよりもDOやTOで大きくなるが、これは零点振動の大きさの違いに由来するものであると考えられる。
H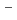 and FH
and FH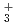
鈴木 机倫*; 石橋 宏章*; 八木 清*; 志賀 基之; 立川 仁典*
Progress in Theoretical Chemistry and Physics, 26, p.207 - 216, 2012/08
Zundel型イオンは特異的に強い水素結合を持つが、その詳細な構造は知られていない。本研究では、第一原理経路積分分子動力学シミュレーションを用いて、フッ素のZundel型イオンであるFHイオン, FHイオンとその重水素置換体の構造について調べた。これらのイオンの水素結合において、H/D原子は、原子核の量子性のため、F原子間の中心のまわりで大きく構造がゆらいでいることがわかった。また、FHやFHのFHやFF原子間距離は、その重水素置換体のFDやFDのものよりも長くなる。この結果について、他のZundel型イオンOH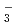 , NH
, NH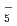 , OH
, OH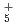 , NH
, NH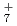 と比較したところ、それらの重原子間の平均距離とその重水素置換効果に相関があることを発見した。なお、FHについては、実験結果や、振動配置間相互作用法による計算結果とよく一致していることがわかり、用いた手法の精度が極めて高いことが立証できた。本成果は、水素結合等での同位体効果やその量子性などを考慮する第一原理計算手法の開発とその検証にあたり、さまざまな分野での応用が期待できる。
と比較したところ、それらの重原子間の平均距離とその重水素置換効果に相関があることを発見した。なお、FHについては、実験結果や、振動配置間相互作用法による計算結果とよく一致していることがわかり、用いた手法の精度が極めて高いことが立証できた。本成果は、水素結合等での同位体効果やその量子性などを考慮する第一原理計算手法の開発とその検証にあたり、さまざまな分野での応用が期待できる。
吉川 武宏*; 菅原 修一*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Chemical Physics, 394(1), p.46 - 51, 2012/02
被引用回数:18 パーセンタイル:55.59(Chemistry, Physical)プロトン移動反応は、水や生体内で起こる基本的反応であり、ポルフィセン分子は、それが協同的に起こる二重プロトン移動反応を示す典型的な系の一として多くの研究者の注目を受けている。本研究では、このポルフィセン分子における二重プロトン移動反応における同位体置換効果を調べるため、経路積分分子動力学シミュレーションを行った。その結果、室温において、ポルフィセンの二重プロトン移動は、おもに協同的機構で起こり、二次の鞍点の遷移状態を通過することで反応が進むことがわかった他、同位体置換は反応機構に大きな影響を与え、温度が上がると逐次反応の寄与が増えることも判明した。さらに、この際、面外の分子振動は、協同的機構を抑える働きをしていることもわかり、反応メカニズムの概略を理解することに成功した。これらの研究成果は、化学反応の基礎的理解を進めるものと位置付けられる一方、原子・分子のシミュレーション技術をより高精度にするための必須な技術開発である。
O)小泉 亮人*; 鈴木 机倫*; 志賀 基之; 立川 仁典*
International Journal of Quantum Chemistry, 112(1), p.136 - 139, 2012/01
被引用回数:0 パーセンタイル:0.05(Chemistry, Physical)本論文発表では、金属水酸化物MOH(HO)クラスターの構造と動力学について、第一原理経路積分分子動力学法による計算機シミュレーションを行った成果について報告する。対象とした系では、アルカリ金属類(M=Li, Na, K)の場合は、Mイオンの移動と協同して、水酸化物イオンと水分子の間でプロトン移動が起きるが、これに反して、貴金属類(M=Cu, Ag, Au)の場合は、このようなプロトン移動プロセスは全く見られなかった。これは、貴金属と水の間の結合の方位性がプロトン移動の反応障壁を高くしているためであると考えられ、金属水酸化物クラスターに対する新たな知見と位置付けられる。なお、本成果は、科学研究費補助金研究の一環として実施され、原子力材料シミュレーションの高度化手法の金属水酸化物クラスターへ応用例であり、基礎化学の発展に資する成果である。
杉本 昌崇*; 志賀 基之; 立川 仁典*
Computational and Theoretical Chemistry, 975(1-3), p.31 - 37, 2011/11
被引用回数:5 パーセンタイル:11.26(Chemistry, Physical)本論文では、原子核の量子統計的ゆらぎを考慮した第一原理分子動力学シミュレーションを用いて、一連の水素クラスター陽イオンH, H, H, H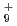 の解析を行った。その結果,クラスターの解離反応H
の解析を行った。その結果,クラスターの解離反応H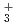 (H
(H )
)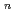
 H(H)
H(H)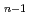 +Hに必要なエネルギーが5.4, 2.1, 3.2kcal/molと見積もられ、実験値と定性的に一致した結果が得られた。解析の結果、水素クラスター解離過程は、量子的な零点振動とその非調和性の影響を強く受けることがわかった。以上、本計算手法を用いると、同位体を有する化合物の化学反応における同位体効果が計算可能であり、原子力材料シミュレーション研究の高度化に資する成果である。
+Hに必要なエネルギーが5.4, 2.1, 3.2kcal/molと見積もられ、実験値と定性的に一致した結果が得られた。解析の結果、水素クラスター解離過程は、量子的な零点振動とその非調和性の影響を強く受けることがわかった。以上、本計算手法を用いると、同位体を有する化合物の化学反応における同位体効果が計算可能であり、原子力材料シミュレーション研究の高度化に資する成果である。
大道 雅史*; 小泉 亮人*; 志賀 基之; 立川 仁典*
Theoretical Chemistry Accounts, 130(2-3), p.385 - 391, 2011/10
被引用回数:6 パーセンタイル:13.82(Chemistry, Physical)本論文発表では、Watson-Crickタイプのグアニン-シトシン塩基対の水素結合構造について、半経験的ポテンシャルエネルギー面を用いた古典的なハイブリッドモンテカルロ法及び量子的な経路積分ハイブリッドモンテカルロ法で計算した成果について報告する。得られた成果としては、3つのNH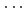 X水素結合部を詳しく調べたところ、量子効果によって分子内NH結合が伸び、HXが縮む傾向が見られたことである。ここで、水素結合距離NXは、HX距離とは相関するが、NH距離とは相関しないこと、また、隣同士の水素結合距離には相関があることもわかった。なお、本研究は科学研究費補助金研究の一環として実施された研究であり、原子力材料物性シミュレーションの高度化手法の生体分子への適用により得られた成果である。
X水素結合部を詳しく調べたところ、量子効果によって分子内NH結合が伸び、HXが縮む傾向が見られたことである。ここで、水素結合距離NXは、HX距離とは相関するが、NH距離とは相関しないこと、また、隣同士の水素結合距離には相関があることもわかった。なお、本研究は科学研究費補助金研究の一環として実施された研究であり、原子力材料物性シミュレーションの高度化手法の生体分子への適用により得られた成果である。
菅原 修一*; 吉川 武宏*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Journal of Physical Chemistry A, 115(42), p.11486 - 11494, 2011/09
被引用回数:19 パーセンタイル:56.05(Chemistry, Physical)本論文発表では、硫酸水和物クラスターにおけるイオン解離や水素結合交替プロセスを明らかにするため、量子的な経路積分分子動力学シミュレーションを行った成果について報告する。得られた成果は、クラスターサイズが大きくなるにつれ、硫酸の第1,第2脱プロトン化が起こりやすくなることがわかったほか、脱プロトン化過程の反応機構において、プロトン移動に関与する硫酸の酸素と水の酸素の間の距離が短くなるという知見である。これらの結果から、プロトンを受容する水の周りの配位数が重要な役割を担っていることが結論づけられる。本研究は、科学研究費補助金研究の一環として行われ、原子力材料物性シミュレーションの高度化手法を硫酸水和クラスターに応用することで得られた成果であり、基礎化学の発展に資する成果と位置付けられる。
小泉 亮人*; 鈴木 机倫*; 志賀 基之; 立川 仁典*
Journal of Chemical Physics, 134(3), p.031101_1 - 031101_3, 2011/01
被引用回数:14 パーセンタイル:42.14(Chemistry, Physical)本論文では、開発した原子核の量子効果を考慮する第一原理計算手法を、水素結合を有する複数の化合物に応用した場合に得られた成果について発表する。なお、開発当該手法の特徴は、第一原理計算に、経路積分により原子核の量子効果を付加することで、より正確に化合物のダイナミクスが再現可能になった点にあり、その手法の適用例として恰好の対象となる同位体異性化合物のMH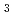 O
O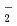 とMDOに対して、各々、M
とMDOに対して、各々、M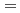 Li, Na, Kとし、そのダイナミクスを比較することで、正イオンの並進運動とプロトン移動の連成が実現していることがわかった。また、小さな正イオンほど、MHOの構造をより大きく歪ませるため、プロトン移動の障壁が高くなり、振動励起エネルギーが低くなることも判明した。これらの成果から、当該手法を用いると、従来、困難とされた水素同位体を含む化合物の詳細な構造解析が可能であることがわかる。以上、本研究成果によりさまざまな同位体を有する化合物の化学反応における同位体効果の理解が進展する。
Li, Na, Kとし、そのダイナミクスを比較することで、正イオンの並進運動とプロトン移動の連成が実現していることがわかった。また、小さな正イオンほど、MHOの構造をより大きく歪ませるため、プロトン移動の障壁が高くなり、振動励起エネルギーが低くなることも判明した。これらの成果から、当該手法を用いると、従来、困難とされた水素同位体を含む化合物の詳細な構造解析が可能であることがわかる。以上、本研究成果によりさまざまな同位体を有する化合物の化学反応における同位体効果の理解が進展する。
吉川 武宏*; 菅原 修一*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Chemical Physics Letters, 496(1-3), p.14 - 19, 2010/08
被引用回数:27 パーセンタイル:67.43(Chemistry, Physical)全次元の経路積分分子動力学シミュレーションにより、ポルフィセンのプロトン移動による互変異性化が協奏的か逐次的かを調べた。その結果、二重プロトン移動は二次の鞍点構造を経る協奏的反応経路を通りやすいが、温度が高くなるにつれ、逐次的な機構の寄与が増すことがわかった。核の量子効果がプロトン移動メカニズムを決めるのに重要な役割を果たしている。
 path integral hybrid Monte Carlo based on the fourth-order Trotter expansion; Application to fluoride ion-water cluster
path integral hybrid Monte Carlo based on the fourth-order Trotter expansion; Application to fluoride ion-water cluster鈴木 机倫*; 立川 仁典*; 志賀 基之
Journal of Chemical Physics, 132(14), p.144108_1 - 144108_7, 2010/04
被引用回数:34 パーセンタイル:75.25(Chemistry, Physical)分子シミュレーションの1つの有効な新手法として、四次トロッター分解に基づいた効率的な経路積分ハイブリッドモンテカルロ法(PIHMC)を提案する。すなわち、二次の有効的な力を用いて短い試行トラジェクトリを生成することで計算のかかるヘシアン行列を避けつつ、最終的なアクセプタンスは四次の有効ポテンシャルで決めるというものである。計算効率について、標準的な2次,4次のPIHMCや経路積分分子動力学法(PIMD)と比較する。この方法を用いて、室温でのフッ素水イオン系の第一原理PIHMC計算を行い、その幾何学的同位体効果について焦点を当てて議論する。
 path integral simulation
path integral simulation志賀 基之; 鈴木 机倫*; 立川 仁典*
Journal of Chemical Physics, 132(11), p.114104_1 - 114104_7, 2010/03
被引用回数:17 パーセンタイル:50.44(Chemistry, Physical)脱プロトン化された水二量体H O
O の化学シフトについて、第一原理経路積分シミュレーションを行った。このシミュレーションから水素結合性のプロトンの等方的遮蔽定数が温度とともに上昇することを予測される。その温度変化率は水素結合を作らないプロトンに比べて一桁大きい。これは低障壁水素結合におけるプロトンの量子的分布が高温と低温で著しく変化する結果から説明される。
の化学シフトについて、第一原理経路積分シミュレーションを行った。このシミュレーションから水素結合性のプロトンの等方的遮蔽定数が温度とともに上昇することを予測される。その温度変化率は水素結合を作らないプロトンに比べて一桁大きい。これは低障壁水素結合におけるプロトンの量子的分布が高温と低温で著しく変化する結果から説明される。
SO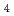
 (HO) (=1-6) on semiempirical PM6 potential surfaces
(HO) (=1-6) on semiempirical PM6 potential surfaces柿崎 陽*; 茂木 春樹*; 吉川 武宏*; 高柳 敏幸*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
半経験的PM6法を用いて、一連の硫酸クラスターを対象とした量子的な経路積分分子動力学シミュレーションを行った。クラスターサイズが大きくなるほど、イオン解離を起こしやすくなり、コンタクトイオンペアと呼ばれる構造が支配的となる。クラスター構造やプロトン移動過程において原子核の量子効果が重要であることを示す。
O) on a semiempirical potential energy surface高柳 敏幸*; 高橋 健太*; 柿崎 陽*; 志賀 基之; 立川 仁典*
Chemical Physics, 358(3), p.196 - 202, 2009/04
被引用回数:16 パーセンタイル:48.94(Chemistry, Physical)塩酸クラスターHCl(HO)について、半経験的PM3-MAIS分子軌道計算から基底状態のポテンシャル面を求め、経路積分分子動力学シミュレーションを行った。計算の結果、300K以上の温度では構造変化を起こし、液体的な振るまいが見られた。この構造変化では、水素原子核の量子揺らぎが重要な役割を担っている。
O)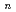 (=2-7) clusters on semiempirical PM6 potential energy surfaces
(=2-7) clusters on semiempirical PM6 potential energy surfaces高柳 敏幸*; 吉川 武宏*; 柿崎 陽*; 志賀 基之; 立川 仁典*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
半経験的分子軌道法であるPM6法に基づいた分子動力学法により、グリシン-水クラスターの熱力学的構造を調べた。クラスターサイズが大きくなるにつれグリシンが中性分子であるよりも双性イオンであるほうが安定になる傾向が見られた。また、中性分子と双性イオンの形での水和構造の違いについて議論する。
鈴木 机倫*; 志賀 基之; 立川 仁典*
Journal of Chemical Physics, 129(144), p.144310_1 - 144310_8, 2008/10
被引用回数:47 パーセンタイル:84.15(Chemistry, Physical)4次トロッター展開による経路積分分子動力学法により、水ダイマーアニオン(HO, DO, TO)の同位体効果について50K 600Kの温度範囲で調べた。200K以下の低い温度では、水素結合を担う水素が二つの酸素と同時に結合したO..
600Kの温度範囲で調べた。200K以下の低い温度では、水素結合を担う水素が二つの酸素と同時に結合したO..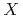 ..O(ここに=H,D,T)という状態を取るが、400K以上の高い温度では、水素がより非局在化してO..-OとO-..Oのような構造が共存する傾向がみられた。低温条件ではH体, D体, T体の順に重くなるほど平均酸素間距離は短くなるが、高温条件ではこの順番が逆転する。この現象を理解するにあたり、O-O伸縮モードと水素移動モードの間のカップリングが重要であると結論づけられる。
..O(ここに=H,D,T)という状態を取るが、400K以上の高い温度では、水素がより非局在化してO..-OとO-..Oのような構造が共存する傾向がみられた。低温条件ではH体, D体, T体の順に重くなるほど平均酸素間距離は短くなるが、高温条件ではこの順番が逆転する。この現象を理解するにあたり、O-O伸縮モードと水素移動モードの間のカップリングが重要であると結論づけられる。
H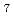
 cation and NH
cation and NH anion by path integral molecular dynamics simulation
anion by path integral molecular dynamics simulation石橋 宏章*; 林 愛子*; 志賀 基之; 立川 仁典*
ChemPhysChem, 9(3), p.383 - 387, 2008/02
被引用回数:24 パーセンタイル:64.37(Chemistry, Physical)第一原理経路積分分子動力学計算により、NHカチオン及びNHアニオンの幾何学的同位体効果について調べた。その結果、これらのイオンの水素結合の構造について、原子核の量子効果が重要な役割を果たすことを見いだした。水素結合を担う水素は、NHでは二つの窒素原子の中心付近にあるのに対し、NDでは一つの窒素側に偏ったところにある。一方、NHとNDともに、水素は一つの窒素側に偏って存在することがわかった。
quantum mechanical/molecular mechanical molecular dynamics using multiple-time-scale approach and perturbation theory志賀 基之; 立川 仁典*
Molecular Simulation, 33(1-2), p.174 - 184, 2007/02
第一原理量子古典分子動力学法は法による非経験的な力場計算と経験的な力場計算を組合せたハイブリッド法で、凝集分子系に対して有効なシミュレーション手法として最近注目を集めている。本研究では、第一原理量子古典分子動力学法を用いて熱力学的解析を行うのに適した手法を提案し、その計算効率や計算精度について議論する。また、この手法の有用性を確かめるため、水溶液の構造とエネルギーの計算結果を報告する。
 path integral molecular dynamics simulation
path integral molecular dynamics simulation林 愛子*; 志賀 基之; 立川 仁典*
Molecular Simulation, 33(1-2), p.185 - 188, 2007/02
被引用回数:3 パーセンタイル:9.21(Chemistry, Physical)近年、二水素結合やリチウム-水素結合など新しいタイプの水素結合様式が実験的に見いだされているが、これらの水素結合の性質や構造は詳細にわかっていないものも多い。これらに対し、第一原理経路積分分子動力学法を用いてCHLiH分子クラスターの計算機シミュレーションを行った結果、これらの結合においても通常の水素結合と同様に大きな同位体効果があることがわかった。
朽津 敬史*; 立川 仁典*; 志賀 基之
Chemical Physics Letters, 433(1-3), p.193 - 198, 2006/12
被引用回数:1 パーセンタイル:2.52(Chemistry, Physical)パルスレーザーにより電子状態を制御すると、通常の状態とは異なる多様なダイナミクスが見られるが、この現象のシミュレーションを行うためにはアト秒からフェムト秒の間の電子状態の変化を追跡することが必要で、その技術はまだあまり進歩していない。本研究では、その候補として電子状態を動的なガウス基底関数の重ね合わせで記述することによって第一原理的に計算する手法を述べ、その手法により水素原子やヘリウム原子が急激に変化する電場の中でどう応答するかを数値的に観察した。
