


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Suzuki, Motofumi*; Tsukamoto, Takashi*; Inoue, Haruhiko*; Watanabe, Satoshi; Matsuhashi, Shimpei; Takahashi, Michiko*; Nakanishi, Hiromi*; Mori, Satoshi*; Nishizawa, Naoko*
Plant Molecular Biology, 66(6), p.609 - 617, 2008/04
Times Cited Count:138 Percentile:95.26(Biochemistry & Molecular Biology)Yamasaki, Chisato*; Murakami, Katsuhiko*; Fujii, Yasuyuki*; Sato, Yoshiharu*; Harada, Erimi*; Takeda, Junichi*; Taniya, Takayuki*; Sakate, Ryuichi*; Kikugawa, Shingo*; Shimada, Makoto*; et al.
Nucleic Acids Research, 36(Database), p.D793 - D799, 2008/01
Times Cited Count:51 Percentile:71.25(Biochemistry & Molecular Biology)Here we report the new features and improvements in our latest release of the H-Invitational Database, a comprehensive annotation resource for human genes and transcripts. H-InvDB, originally developed as an integrated database of the human transcriptome based on extensive annotation of large sets of fulllength cDNA (FLcDNA) clones, now provides annotation for 120 558 human mRNAs extracted from the International Nucleotide Sequence Databases (INSD), in addition to 54 978 human FLcDNAs, in the latest release H-InvDB. We mapped those human transcripts onto the human genome sequences (NCBI build 36.1) and determined 34 699 human gene clusters, which could define 34 057 protein-coding and 642 non-protein-coding loci; 858 transcribed loci overlapped with predicted pseudogenes.
Ishimaru, Yasuhiro*; Kim, S.*; Tsukamoto, Takashi*; Oki, Hiroyuki*; Kobayashi, Takanori*; Watanabe, Satoshi; Matsuhashi, Shimpei; Takahashi, Michiko*; Nakanishi, Hiromi*; Mori, Satoshi*; et al.
Proceedings of the National Academy of Sciences of the United States of America, 104(18), p.7373 - 7378, 2007/05
Times Cited Count:130 Percentile:93.37(Multidisciplinary Sciences)Yura, Kei; Shionyu, Masafumi*; Hagino, Kei*; Hijikata, Atsushi*; Hirashima, Yoshinori*; Nakahara, Taku*; Eguchi, Tatsuya*; Shinoda, Kazuki*; Yamaguchi, Akihiro*; Takahashi, Kenichi*; et al.
Gene, 380(2), p.63 - 71, 2006/10
Times Cited Count:55 Percentile:72.3(Genetics & Heredity)Alternative splicing is a molecular mechanism that produces multiple proteins from a single gene, and is thought to produce variety in proteins translated from a limited number of genes. Here we analyzed how alternative splicing produced variety in protein structure and function, by using human full-length cDNAs, on the assumption that all of the alternatively spliced mRNAs were translated to proteins. We found that the length of alternatively spliced amino acid sequences, in most cases, fell into a size shorter than that of average protein domain. We evaluated comprehensively the presumptive three-dimensional structures of the alternatively spliced products to assess the impact of alternative splicing on gene function. We found that more than half of the products encoded proteins which were involved in signal transduction, transcription and translation, and more than half of alternatively spliced regions comprised interaction sites between proteins and their binding partners, including substrates, DNA/RNA, and other proteins. Intriguingly, 67% of the alternatively spliced isoforms showed significant alterations to regions of the protein structural core, which likely resulted in large conformational change. Based on those findings, we speculate that there are a large number of cases that alternative splicing modulates protein networks through significant alteration in protein conformation.
Suzuki, Motofumi*; Takahashi, Michiko*; Tsukamoto, Takashi*; Watanabe, Satoshi; Matsuhashi, Shimpei; Yazaki, Junshi*; Kishimoto, Naoki*; Kikuchi, Shoshi*; Nakanishi, Hiromi*; Mori, Satoshi*; et al.
Plant Journal, 48(1), p.85 - 97, 2006/10
Times Cited Count:172 Percentile:95.77(Plant Sciences)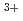 -phytosiderophore and as Fe
-phytosiderophore and as Fe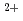
Ishimaru, Yasuhiro*; Suzuki, Motofumi*; Tsukamoto, Takashi*; Suzuki, Kazumasa*; Nakazono, Mikio*; Kobayashi, Takanori*; Wada, Yasuaki*; Watanabe, Satoshi; Matsuhashi, Shimpei; Takahashi, Michiko*; et al.
Plant Journal, 45(3), p.335 - 346, 2006/02
Times Cited Count:545 Percentile:99.64(Plant Sciences)Ichimasa, Michiko*; Awagakubo, Sayuri*; Takahashi, Miho*; Tauchi, Hiroshi*; Hayashi, Takumi; Kobayashi, Kazuhiro; Nishi, Masataka; Ichimasa, Yusuke*
Fusion Science and Technology, 48(1), p.759 - 762, 2005/07
Times Cited Count:7 Percentile:44.9(Nuclear Science & Technology)There exists various kinds of HT oxidizing soil bacteria in the world, and we have conducted the investigation of HT oxidation activity of such bacteria. In the fusion facility where deuterium and tritium will be used as its fuel, the system is necessary to eliminate tritium from atmospheric air. General tritium elimination method is oxidation and dehumidification, and high temperature catalyst is used in the present system for oxidation. Application of the HT oxidation bioreactor, which can oxidize in room temperature, to this oxidation process has possibility to get higher tritium elimination efficiency, so we started to study the bioreactor. In the recent study, we can get high oxidation ratio of 85% in the processing conditions of 200 Bq/cm as tritium concentration in air, 100 cm/min as flow rate and once-through processing using the Caisson Assembly for Tritium Safety Study (CATS) in JAERI. This result encourages this development study.
as tritium concentration in air, 100 cm/min as flow rate and once-through processing using the Caisson Assembly for Tritium Safety Study (CATS) in JAERI. This result encourages this development study.
Shionyu, Masafumi*; Yura, Kei; Hijikata, Atsushi*; Nakahara, Taku*; Shinoda, Kazuki*; Yamaguchi, Akihiro*; Takahashi, Kenichi*; Go, Michiko*
no journal, ,
Alternative splicing (AS) is a cellular process where multiple mature mRNAs are produced from a single gene by different usage of exons. From computational and experimental approaches, it is estimated that 30-70% of human genes undergo alternative splicing. There are a number of reports on spliced mRNAs involved in biological processes, yet functional analyses of a large number of proteins produced by AS remain to be performed. Functional analyses of these proteins by experiments are time-consuming, and therefore, a computational approach that can estimate effect of AS on proteins is required. We have developed a pipeline that can systematically detect AS regions, which are defined as protein regions modified by alternatively spliced exon, using genomic sequences and full-length transcripts data. The pipeline further assigned AS regions to protein three-dimensional structures and can estimate effects of AS on protein conformation stability and functional sites. We analyzed human AS regions using our pipeline and found that about half of AS regions fell into a size shorter than 100 amino acid residues. We, then, assessed the relationship between AS regions and protein structural domains and found that about 40% of AS regions were placed within a domain and the ratio of AS regions corresponding to domains was only about 10%. This result suggests that AS regulates protein function through alteration of segments within a domain rather than through switching protein domains. We will discuss how AS regulates protein function through alteration of segments within a domain.
