


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Nakagawa, Hiroshi; Tamada, Taro*
Frontiers in Chemistry (Internet), 9, p.738077_1 - 738077_7, 2021/10
Times Cited Count:10 Percentile:61.19(Chemistry, Multidisciplinary)Protein hydration is crucial for the stability and molecular recognition of a protein. Water molecules interact with a protein surface via hydrogen bonding. Here, we examined the hydration structure and hydrogen bonding state of a globular protein, staphylococcal nuclease, at various hydration levels in a crystalline state by all-atom molecular dynamics simulation. The hydrophobic residue surface was found to be more hydrated than the hydrophilic residue surface, but both were uniformly hydrated in response to increased water content. In addition, the hydrogen bonds in hydrated water have a tetrahedral structure, which is not much different from the structure of bulk water. The hydrogen bonding structure is compatible with the results of neutron crystallography. The simulations are useful for analyzing the hydration structure and hydrogen bonding state in the crystalline state, and will greatly assist in the further analysis of the information obtained from crystal structure analysis.
Tashiro, Koji*; Kusaka, Katsuhiro*; Hosoya, Takaaki*; Ohara, Takashi; Hanesaka, Makoto*; Yoshizawa, Yoshinori*; Yamamoto, Hiroko*; Niimura, Nobuo*; Tanaka, Ichiro*; Kurihara, Kazuo*; et al.
Macromolecules, 51(11), p.3911 - 3922, 2018/06
Times Cited Count:5 Percentile:17.66(Polymer Science) binding site of a
binding site of a 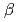 -lactamase from the halophile by anomalous X-ray diffraction
-lactamase from the halophile by anomalous X-ray diffractionArai, Shigeki; Shibazaki, Chie; Shimizu, Rumi; Adachi, Motoyasu; Tamada, Taro; Tokunaga, Hiroko*; Ishibashi, Matsujiro*; Tokunaga, Masao*; Kuroki, Ryota
Kyushu Shinkurotoronko Kenkyu Senta Nempo, 2014, p.17 - 19, 2016/03
no abstracts in English
Tamada, Taro; Shinmi, Daisuke*; Ikeda, Masahiro*; Yonezawa, Yasushi*; Kataoka, Shiro*; Kuroki, Ryota; Mori, Eiji*; Motoki, Kazuhiro*
Scientific Reports (Internet), 5, p.17936_1 - 17936_12, 2015/12
Times Cited Count:25 Percentile:62.95(Multidisciplinary Sciences)The fully human monoclonal antibody KMTR2 acts as a strong direct agonist for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor 2 (TRAIL-R2), which is capable of inducing apoptotic cell death without cross-linking. To investigate the mechanism of direct agonistic activity induced by KMTR2, the crystal structure of the extracellular region of TRAIL-R2 and a Fab fragment derived from KMTR2 (KMTR2-Fab) was determined to 2.1 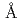 resolution. Two KMTR2-Fabs assembled with the complementarity-determining region 2 of the light chain via two-fold crystallographic symmetry, suggesting that the KMTR2-Fab assembly tended to enhance TRAIL-R2 oligomerization. A single mutation at Asn53 to Arg located at the two-fold interface in the KMTR2 resulted in a loss of its apoptotic activity, although it retained its antigen-binding activity. These results indicate that the strong agonistic activity, such as apoptotic signaling and tumor regression, induced by KMTR2 is attributed to TRAIL-R2 superoligomerization induced by the interdimerization of KMTR2.
resolution. Two KMTR2-Fabs assembled with the complementarity-determining region 2 of the light chain via two-fold crystallographic symmetry, suggesting that the KMTR2-Fab assembly tended to enhance TRAIL-R2 oligomerization. A single mutation at Asn53 to Arg located at the two-fold interface in the KMTR2 resulted in a loss of its apoptotic activity, although it retained its antigen-binding activity. These results indicate that the strong agonistic activity, such as apoptotic signaling and tumor regression, induced by KMTR2 is attributed to TRAIL-R2 superoligomerization induced by the interdimerization of KMTR2.
Unno, Masayoshi*; Sugishima, Masakazu*; Wada, Kei*; Hagiwara, Yoshinori*; Kusaka, Katsuhiro*; Tamada, Taro; Fukuyama, Keiichi*
Nihon Kessho Gakkai-Shi, 57(5), p.297 - 303, 2015/10
Bilin compounds are fundamentally important for oxygenic photosynthetic organisms, because they are utilized as pigments for photosynthesis (phycobilins) and photoreceptors (phytochromobilin). Phycocyanobilin (PCB), a phycobilin, comprises the chromophore of algal phytochromes and the core phycobiliprotein antennae of cyanobacteria and red algae. PCB is biosynthesized by a member of the ferredoxin-dependent bilin reductase family, phycocyanobilin:ferredoxin oxidoreductase (PcyA). In the present study, we determined the neutron crystal structure of PcyA in complex with its substrate biliverdin (BV). This neutron structure revealed the protonation state of BV and the surrounding residues. We found that two forms of BV, neutral BV and protonated BVH, were coupled with the two conformation/protonation states of the essential residue Asp105. Further, His88 and His74 near BV were singly protonated and were connected with an intervening hydronium ion. Neutron analysis also revealed how X-ray irradiation of the PcyA-BV crystal altered the structure of the PcyA-BV complex.
Tomoyori, Katsuaki; Kurihara, Kazuo; Tamada, Taro; Kuroki, Ryota
JPS Conference Proceedings (Internet), 8, p.036004_1 - 036004_6, 2015/09
We aim to build a high-resolution neutron time-of-flight diffractometer for biomacromolecules at the Materials and Life Science Experimental Facility (MLF) at the Japan Proton Accelerator Research Complex (J-PARC) that allows the collection of neutron diffraction data from crystals with unit cells of 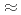 250 ;. Considering both the flux and pulse width necessary to realize data collection covering a minimum d-spacing of 2.0 ; and with a unit cell constant of 250 ; we chose a decoupled moderator (DM) as the appropriate source for this high-resolution diffractometer. We considered a simple instrumentation model that includes a moderator, neutron guide, sample size, and neutron detector; we then investigated its spot separation performance and estimated the instrumental parameters for the design of a new diffractometer for protein crystals with large unit cells at J-PARC/MLF. It is preferable to extend the total flight path to resolve Bragg reflections for protein crystals with large unit cells as the scattering angle increases. Meanwhile, to ensure resolvable detection capacity at the middle scattering angle region (2
250 ;. Considering both the flux and pulse width necessary to realize data collection covering a minimum d-spacing of 2.0 ; and with a unit cell constant of 250 ; we chose a decoupled moderator (DM) as the appropriate source for this high-resolution diffractometer. We considered a simple instrumentation model that includes a moderator, neutron guide, sample size, and neutron detector; we then investigated its spot separation performance and estimated the instrumental parameters for the design of a new diffractometer for protein crystals with large unit cells at J-PARC/MLF. It is preferable to extend the total flight path to resolve Bragg reflections for protein crystals with large unit cells as the scattering angle increases. Meanwhile, to ensure resolvable detection capacity at the middle scattering angle region (2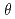 90
90 ), it is necessary to restrict the angular divergence. In the case of
), it is necessary to restrict the angular divergence. In the case of  0.2, scattering angles from around 290 to higher backscattering angles are more efficient for protein crystals with large unit cells (
0.2, scattering angles from around 290 to higher backscattering angles are more efficient for protein crystals with large unit cells (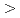 250 ) with a resolution of 2.0 .
250 ) with a resolution of 2.0 .
Hirano, Yu; Kimura, Shigenobu*; Tamada, Taro
Acta Crystallographica Section D, 71(7), p.1572 - 1581, 2015/07
Times Cited Count:1 Percentile:11.24(Biochemical Research Methods)Mammalian microsomal cytochrome has multiple electron-transfer partners that function in various electron-transfer reactions. Four crystal structures of the solubilized haem-binding domain of cytochrome from porcine liver were determined at sub-angstrom resolution (0.76-0.95 ) in two crystal forms for both the oxidized and reduced states. The high-resolution structures clearly displayed the electron density of H atoms in some amino-acid residues. Unrestrained refinement of bond lengths revealed that the protonation states of the haem propionate group may be involved in regulation of the haem redox properties. The haem Fe coordination geometry did not show significant differences between the oxidized and reduced structures. However, structural differences between the oxidized and reduced states were observed in the hydrogen-bond network around the axial ligand His68. The hydrogen-bond network could be involved in regulating the redox states of the haem group.
Unno, Masayoshi*; Ishikawa, Kumiko*; Kusaka, Katsuhiro*; Tamada, Taro; Hagiwara, Yoshinori*; Sugishima, Masakazu*; Wada, Kei*; Yamada, Taro*; Tomoyori, Katsuaki; Hosoya, Takaaki*; et al.
Journal of the American Chemical Society, 137(16), p.5452 - 5460, 2015/04
Times Cited Count:28 Percentile:63.97(Chemistry, Multidisciplinary)Phycocyanobilin, a light-harvesting and photoreceptor pigment in higher plants, algae, and cyanobacteria, is synthesized from biliverdin IX (BV) by phycocyanobilin:ferredoxin oxidoreductase (PcyA) via two steps of two-proton-coupled two-electron reduction. We determined the neutron structure of PcyA from cyanobacteria complexed with BV, revealing the exact location of the hydrogen atoms involved in catalysis. Notably, approximately half of the BV bound to PcyA was BVH, a state in which all four pyrrole nitrogen atoms were protonated. The protonation states of BV complemented the protonation of adjacent Asp105. The "axial "water molecule that interacts with the neutral pyrrole nitrogen of the A-ring was identified. His88 N
(BV) by phycocyanobilin:ferredoxin oxidoreductase (PcyA) via two steps of two-proton-coupled two-electron reduction. We determined the neutron structure of PcyA from cyanobacteria complexed with BV, revealing the exact location of the hydrogen atoms involved in catalysis. Notably, approximately half of the BV bound to PcyA was BVH, a state in which all four pyrrole nitrogen atoms were protonated. The protonation states of BV complemented the protonation of adjacent Asp105. The "axial "water molecule that interacts with the neutral pyrrole nitrogen of the A-ring was identified. His88 N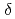 was protonated to form a hydrogen bond with the lactam O atom of the BV A-ring. His88 and His74 were linked by hydrogen bonds via H
was protonated to form a hydrogen bond with the lactam O atom of the BV A-ring. His88 and His74 were linked by hydrogen bonds via H O. These results imply that Asp105, His88, and the axial water molecule contribute to proton transfer during PcyA catalysis.
O. These results imply that Asp105, His88, and the axial water molecule contribute to proton transfer during PcyA catalysis.
-lactamase from the moderate halophile 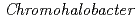 sp.560 and the discovery of a Cs-selective binding site
sp.560 and the discovery of a Cs-selective binding siteArai, Shigeki; Yonezawa, Yasushi*; Okazaki, Nobuo*; Matsumoto, Fumiko*; Shibazaki, Chie; Shimizu, Rumi; Yamada, Mitsugu*; Adachi, Motoyasu; Tamada, Taro; Kawamoto, Masahide*; et al.
Acta Crystallographica Section D, 71(3), p.541 - 554, 2015/03
Times Cited Count:7 Percentile:50.44(Biochemical Research Methods)The crystal structure of halophilic -lactamase from sp.560 (HaBLA) was determined using X-ray crystallography. Moreover, the locations of bound Sr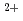 and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-
and Cs ions were identified by anomalous X-ray diffraction. The location of one Cs specific binding site was identified on HaBLA even in the presence of 9-fold molar excess of Na (90 mM Na /10 mM Cs). This Cs binding site is formed by two main-chain O atoms and an aromatic ring of a side chain of Trp. An aromatic ring of Trp interacts with Cs by the cation-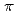 interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
interaction. The observation of a selective and high-affinity Cs binding site provides important information that is useful for designing artificial Cs binding sites useful in bioremediation of radioactive isotopes.
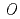 -glucosyltransferase from
-glucosyltransferase from 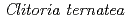
Hiromoto, Takeshi; Honjo, Eijiro*; Noda, Hisanobu*; Tamada, Taro; Kazuma, Kohei*; Suzuki, Masahiko*; Blaber, M.; Kuroki, Ryota
Protein Science, 24(3), p.395 - 407, 2015/03
Times Cited Count:62 Percentile:88.89(Biochemistry & Molecular Biology)UDP-glucose: anthocyanidin 3--glucosyltransferase (UGT78K6) from catalyzes the transfer of glucose from UDP-glucose to anthocyanidins such as delphinidin. To understand the acceptor-recognition scheme of UGT78K6, the crystal structure of UGT78K6 and its complex forms with anthocyanidin delphinidin and petunidin, and flavonol kaempferol were determined to resolutions of 1.85 , 2.55 , 2.70 and 1.75 respectively. The anthocyanidin- and flavonol-acceptor binding details are almost identical in each complex structure, although the glucosylation activities against each acceptor were significantly different. The acceptor substrates in UGT78K6 are reversely bound to its binding site by a 180 rotation about the O1-O3 axis of the flavonoid backbones observed in 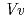 GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
GT1 and UGT78G1. These substrate recognition schemes suggest the potential for controlled synthesis of natural pigments.
Tomoyori, Katsuaki; Hirano, Yu; Kurihara, Kazuo; Tamada, Taro
Journal of Physics; Conference Series, 664, p.072049_1 - 072049_7, 2015/00
Times Cited Count:4 Percentile:81.62(Physics, Nuclear)In neutron protein crystallography, it should be also emphasized that the weak Bragg reflections due to the large unit cells may be buried beneath the strong background caused by the incoherent scattering of hydrogen atoms. Therefore, the background estimation from the source is more reliable to improve the accuracy of Bragg integral intensity. We propose the adoption of Statistics-sensitive Nonlinear Iterative Peak-clipping (SNIP) algorithm for background estimation, which can eliminate the background from the source spectrum as well as statistically enhance low peaks. In addition, it was recently reported that the Landau and Vavilov distributions, which are used to describe the energy loss of charged particles traversing a thin absorber were found to be in excellent agreement with the observed TOF profile. I show that it may be beneficial to establish a profile-fitting method with the combined use of the Landau/Vavilov functions to faithfully reproduce the TOF peak shape and a SNIP background evaluation algorithm to eliminate statistical fluctuations.
Tashiro, Koji*; Hanesaka, Makoto*; Yamamoto, Hiroko*; Wasanasuk, K.*; Jayaratri, P.*; Yoshizawa, Yoshinori*; Tanaka, Ichiro*; Niimura, Nobuo*; Kusaka, Katsuhiro*; Hosoya, Takaaki*; et al.
Kobunshi Rombunshu, 71(11), p.508 - 526, 2014/11
Times Cited Count:6 Percentile:22.16(Polymer Science)The crystal structure analysis of various polymer substances has been reviewed on the basis of wide-angle high-energy X-ray and neutron diffraction data. The progress in structural analytical techniques of polymer crystals have been reviewed at first. The structural models proposed so far were reinvestigated and new models have been proposed for various kinds of polymer crystals including polyethylene, poly(vinyl alcohol), poly(lactic acid) and its stereocomplex etc. The hydrogen atomic positions were also clarified by the quantitative analysis of wide-angle neutron diffraction data, from which the physical properties of polymer crystals have been evaluated theoretically. The bonded electron density distribution has been estimated for a polydiacetylene single crystal on the basis of the so-called X-N method or by the combination of structural information derived from X-ray and neutron diffraction data analysis. Some comments have been added about future developments in the field of structure-property relationship determination.
Tomoyori, Katsuaki; Kusaka, Katsuhiro*; Yamada, Taro*; Tamada, Taro
Journal of Structural and Functional Genomics, 15(3), p.131 - 135, 2014/09
We plan to design a high-resolution biomacromolecule neutron time-of-flight diffractometer, which allows us to collect data from crystals with unit cells above 250 in the Materials and Life Science Experimental Facility at the Japan Proton Accelerator Research Complex. This new diffractometer can be used for a detailed analysis of large proteins such as membrane proteins and supermolecular complex. A quantitative comparison of the intensity and pulse width of a decoupled moderator (DM) against a coupled moderator (CM) considering the pulse width time resolution indicated that the DM satisfies the criteria for our diffractometer rather than the CM. The results suggested that a characteristic feature of the DM, i.e., narrow pulse width with a short tail, is crucial for the separation of Bragg reflections from crystals with large unit cells. On the other hand, it should be noted that the weak signals from the DM are buried under the high-level background caused by the incoherent scattering of hydrogen atoms, especially, in the case of large unit cells. We propose a profile-fitting integration method combined with the energy loss functions and a background subtraction method achieved by employing the Statistics-sensitive Nonlinear Iterative Peak-clipping algorithm.
Kurihara, Kazuo; Tamada, Taro
Hamon, 24(3), p.190 - 199, 2014/08
In order to overcome low flux of neutron sources as well as weak diffraction intensity from bio-macromolecule crystals, several devices have been developed in neutron biological crystallography. Elastically bent Si monochromator has contributed the increase of incident beam intensity, and neutron imaging plate (NIP) has provided large detecting area. In particular, the successful development of the NIP made a breakthrough in this research field. Additionally, recent advances in techniques for cryogenic temperature measurement, growth of large crystal and sample deuteration have made a contribution to efficient measurement performance. Currently, a total of six diffractometers for bio-macromolecule are available at research reactors in the world. Neutron crystallography is on the verge of becoming a prevalent method for structural study on bio-macromolecules.
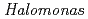 sp.593
sp.593Arai, Shigeki; Yonezawa, Yasushi*; Ishibashi, Matsujiro*; Matsumoto, Fumiko*; Adachi, Motoyasu; Tamada, Taro; Tokunaga, Hiroko*; Blaber, M.; Tokunaga, Masao*; Kuroki, Ryota
Acta Crystallographica Section D, 70(3), p.811 - 820, 2014/03
Times Cited Count:12 Percentile:62.81(Biochemical Research Methods)In order to clarify the structural basis of halophilic characteristics of an alkaline phosphatase derived from the moderate halophile sp.593 (HaAP), the tertiary structure of HaAP was determined to 2.1 resolution by X-ray crystallography. Structural properties of surface negative charge and core hydrophobicity are shown to be intermediate between halophile and non-halophile characteristics, and may explain the unique functional adaptation to a wide-range of salt concentration.
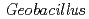 copper nitrite reductase; Involvement of the unique N-terminal region in the interprotein electron transfer with its redox partner
copper nitrite reductase; Involvement of the unique N-terminal region in the interprotein electron transfer with its redox partnerFukuda, Yota*; Koteishi, Hiroyasu*; Yoneda, Ryohei*; Tamada, Taro; Takami, Hideto*; Inoue, Tsuyoshi*; Nojiri, Masaki
Biochimica et Biophysica Acta; Bioenergetics, 1837(3), p.396 - 405, 2014/03
Times Cited Count:15 Percentile:46.83(Biochemistry & Molecular Biology)The crystal structures of copper-containing nitrite reductase (CuNiR) from the thermophilic Gram-positive bacterium 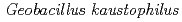 HTA426 and the amino (N)-terminal 68 residue-deleted mutant were determined at resolutions of 1.3 and 1.8, respectively. Both structures show a striking resemblance with the overall structure of the well-known CuNiRs composed of two Greek key -barrel domains; however, a remarkable structural difference was found in the N-terminal region. The unique region has one -strand and one -helix extended to the northern surface of the type-1 copper site. The superposition of the CuNiR model on the electron-transfer complex structure of CuNiR with the redox partner cytochrome
HTA426 and the amino (N)-terminal 68 residue-deleted mutant were determined at resolutions of 1.3 and 1.8, respectively. Both structures show a striking resemblance with the overall structure of the well-known CuNiRs composed of two Greek key -barrel domains; however, a remarkable structural difference was found in the N-terminal region. The unique region has one -strand and one -helix extended to the northern surface of the type-1 copper site. The superposition of the CuNiR model on the electron-transfer complex structure of CuNiR with the redox partner cytochrome 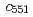 in other denitrifier system led us to infer that this region contributes to the transient binding with the partner protein during the interprotein electron transfer reaction in the system. Furthermore, electron-transfer kinetics experiments using N-terminal residue-deleted mutant and the redox partner, cytochrome , were carried out. These structural and kinetics studies demonstrate that that region is directly involved in the specific partner recognition.
in other denitrifier system led us to infer that this region contributes to the transient binding with the partner protein during the interprotein electron transfer reaction in the system. Furthermore, electron-transfer kinetics experiments using N-terminal residue-deleted mutant and the redox partner, cytochrome , were carried out. These structural and kinetics studies demonstrate that that region is directly involved in the specific partner recognition.
 ; Assignment of bound diatomic molecules as O
; Assignment of bound diatomic molecules as O
Murakawa, Takeshi*; Hayashi, Hideyuki*; Sunami, Tomoko; Kurihara, Kazuo; Tamada, Taro; Kuroki, Ryota; Suzuki, Mamoru*; Tanizawa, Katsuyuki*; Okajima, Toshihide*
Acta Crystallographica Section D, 69(12), p.2483 - 2494, 2013/12
Times Cited Count:14 Percentile:68.53(Biochemical Research Methods)The crystal structure of a Cu amine oxidase from was determined at 1.08 resolution with the use of low-molecular-weight polyethylene glycol (LMW PEG; average molecular weight  200) as a cryoprotectant. The final crystallographic
200) as a cryoprotectant. The final crystallographic 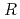 -factor and
-factor and  value are 13.0% and 15.0%, respectively. Several molecules of LMW PEG were found to occupy cavities in the protein interior including the active site, which resulted in the marked reduction of the overall
value are 13.0% and 15.0%, respectively. Several molecules of LMW PEG were found to occupy cavities in the protein interior including the active site, which resulted in the marked reduction of the overall 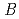 factor and consequently led to a sub-atomic resolution structure for a relatively large protein with a monomer molecular weight of 70,000. About 40% of all the presumed hydrogen atoms were observed as clear electron densities in the
factor and consequently led to a sub-atomic resolution structure for a relatively large protein with a monomer molecular weight of 70,000. About 40% of all the presumed hydrogen atoms were observed as clear electron densities in the 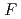
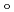 -
- 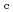 difference map. Multiple minor conformers were also identified for many residues. Anisotropic displacement fluctuations were evaluated in the active site that contains a post-translationally derived quinone cofactor and a Cu atom. Furthermore, diatomic molecules, most likely molecular oxygen, are bound to the protein, one of which is located in the region that has been previously proposed as an entry route for the substrate dioxygen from the central cavity of the dimer interface to the active site.
difference map. Multiple minor conformers were also identified for many residues. Anisotropic displacement fluctuations were evaluated in the active site that contains a post-translationally derived quinone cofactor and a Cu atom. Furthermore, diatomic molecules, most likely molecular oxygen, are bound to the protein, one of which is located in the region that has been previously proposed as an entry route for the substrate dioxygen from the central cavity of the dimer interface to the active site.
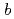
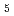 reductase by X-ray crystallography; New insights into regulation of efficient electron transfer
reductase by X-ray crystallography; New insights into regulation of efficient electron transferYamada, Mitsugu*; Tamada, Taro; Takeda, Kazuki*; Matsumoto, Fumiko*; Ono, Hiraku*; Kosugi, Masayuki*; Takaba, Kiyofumi*; Shoyama, Yoshinari*; Kimura, Shigenobu*; Kuroki, Ryota; et al.
Journal of Molecular Biology, 425(22), p.4295 - 4306, 2013/11
Times Cited Count:21 Percentile:50.33(Biochemistry & Molecular Biology)NADH-Cytochrome reductase (b5R), a flavoprotein consisting of NADH and flavin adenine dinucleotide (FAD) binding domains, catalyzes electron transfer from the two-electron carrier NADH to the one-electron carrier cytochrome (Cb5). The crystal structures of both the fully reduced form and the oxidized form of porcine liver b5R were determined. In the reduced b5R structure determined at 1.68 resolution, the relative configuration of the two domains was slightly shifted in comparison with that of the oxidized form. This shift resulted in an increase in the solvent-accessible surface area of FAD and created a new hydrogen-bonding interaction between the N5 atom of the isoalloxazine ring of FAD and the hydroxyl oxygen atom of Thr66, which is considered to be a key residue in the release of a proton from the N5 atom. The isoalloxazine ring of FAD in the reduced form is flat as in the oxidized form and stacked together with the nicotinamide ring of NAD. Determination of the oxidized b5R structure, including the hydrogen atoms, determined at 0.78 resolution revealed the details of a hydrogen-bonding network from the N5 atom of FAD to His49 via Thr66. Both of the reduced and oxidized b5R structures explain how backflow in this catalytic cycle is prevented and the transfer of electrons to one-electron acceptors such as Cb5 is accelerated. Furthermore, crystallographic analysis by the cryo-trapping method suggests that re-oxidation follows a two-step mechanism. These results provide structural insights into the catalytic cycle of b5R.
-glucosyltransferase from Hiromoto, Takeshi; Honjo, Eijiro*; Tamada, Taro; Noda, Hisanobu*; Kazuma, Kohei*; Suzuki, Masahiko*; Kuroki, Ryota
Journal of Synchrotron Radiation, 20(6), p.894 - 898, 2013/11
Times Cited Count:38 Percentile:86.36(Instruments & Instrumentation)Flowers of the butterfly pea () accumulate a group of polyacylated anthocyanins, named ternatins, in their petals. The first step in ternatin biosynthesis is the transfer of glucose from UDP-glucose to anthocyanidins such as delphinidin, a reaction catalyzed in  by UDP-glucose:anthocyanidin 3--glucosyltransferase (
by UDP-glucose:anthocyanidin 3--glucosyltransferase (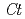 3GT-A; AB185904). To elucidate the structure-function relationship of 3GT-A, recombinant 3GT-A was expressed in
3GT-A; AB185904). To elucidate the structure-function relationship of 3GT-A, recombinant 3GT-A was expressed in 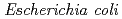 and its tertiary structure was determined to 1.85 , resolution by using X-ray crystallography. The structure of 3GT-A shows a common folding topology, the GT-B fold, comprised of two Rossmann-like // domains and a cleft located between the N- and C-domains containing two cavities that are used as binding sites for the donor (UDP-Glc) and acceptor substrates. By comparing the structure of 3GT-A with that of the flavonoid glycosyltransferase GT1 from red grape (
and its tertiary structure was determined to 1.85 , resolution by using X-ray crystallography. The structure of 3GT-A shows a common folding topology, the GT-B fold, comprised of two Rossmann-like // domains and a cleft located between the N- and C-domains containing two cavities that are used as binding sites for the donor (UDP-Glc) and acceptor substrates. By comparing the structure of 3GT-A with that of the flavonoid glycosyltransferase GT1 from red grape (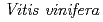 ) in complex with UDP-2-deoxy-2-fluoro glucose and kaempferol, locations of the catalytic His-Asp dyad and the residues involved in recognizing UDP-2-deoxy-2-fluoro glucose were essentially identical in 3GT-A, but certain residues of GT1 involved in binding kaempferol were found to be substituted in 3GT-A. These findings are important for understanding the differentiation of acceptor-substrate recognition in these two enzymes.
) in complex with UDP-2-deoxy-2-fluoro glucose and kaempferol, locations of the catalytic His-Asp dyad and the residues involved in recognizing UDP-2-deoxy-2-fluoro glucose were essentially identical in 3GT-A, but certain residues of GT1 involved in binding kaempferol were found to be substituted in 3GT-A. These findings are important for understanding the differentiation of acceptor-substrate recognition in these two enzymes.
-glucanase from 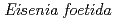
Arimori, Takao*; Ito, Akihiro*; Nakazawa, Masami*; Ueda, Mitsuhiro*; Tamada, Taro
Journal of Synchrotron Radiation, 20(6), p.884 - 889, 2013/11
Times Cited Count:17 Percentile:65.22(Instruments & Instrumentation)The saccharification process is essential for bioethanol production from woody biomass including celluloses. Cold-adapted cellulase, which has sufficient activity at low temperature (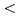 293 K), is capable of reducing heating costs during the saccharification process and is suitable for simultaneous saccharification and fermentation. Endo-1,4--glucanase from the earthworm Eisenia fetida (EF-EG2) belonging to glycoside hydrolase family 9 has been shown to have the highest activity at 313 K, and also retained a comparatively high activity at 283 K. The recombinant EF-EG2 was purified expressed in Pichia pastoris, and then grew needle-shaped crystals with dimensions of 0.02
293 K), is capable of reducing heating costs during the saccharification process and is suitable for simultaneous saccharification and fermentation. Endo-1,4--glucanase from the earthworm Eisenia fetida (EF-EG2) belonging to glycoside hydrolase family 9 has been shown to have the highest activity at 313 K, and also retained a comparatively high activity at 283 K. The recombinant EF-EG2 was purified expressed in Pichia pastoris, and then grew needle-shaped crystals with dimensions of 0.02 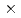 0.02 1 mm. The crystals belonged to the space group P3221 with unit-cell parameters of
0.02 1 mm. The crystals belonged to the space group P3221 with unit-cell parameters of 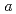 = =136 ,
= =136 , 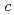 = 55.0 ,. The final model of EF-EG2, including 435 residues, two ions, seven crystallization reagents and 696 waters, was refined to a crystallographic -factor of 14.7% (free -factor of 16.8%) to 1.5 , resolution. The overall structure of EF-EG2 has an (/)
= 55.0 ,. The final model of EF-EG2, including 435 residues, two ions, seven crystallization reagents and 696 waters, was refined to a crystallographic -factor of 14.7% (free -factor of 16.8%) to 1.5 , resolution. The overall structure of EF-EG2 has an (/)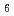 barrel fold which contains a putative active-site cleft and a negatively charged surface. This structural information helps us understand the catalytic and cold adaptation mechanisms of EF-EG2.
barrel fold which contains a putative active-site cleft and a negatively charged surface. This structural information helps us understand the catalytic and cold adaptation mechanisms of EF-EG2.
