


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 -Fe
-FeSuzudo, Tomoaki; Ebihara, Kenichi; Tsuru, Tomohito; Mori, Hideki*
Journal of Applied Physics, 135(7), p.075102_1 - 075102_7, 2024/02
Fracture of body centred cubic (bcc) metals and alloys below the ductile-to-brittle transition temperature is brittle. This is theoretically explained by the notion that the critical stress intensity factor of a given crack front for brittle fracture is smaller than that for plasticdeformation; hence, brittle fracture is chosen over plastic deformation. Although this view is true from a macroscopic point of view, such brittle fracture is always accompanied by small-scale plastic deformation in the vicinity of the crack tip, i.e. crack tip plasticity. This short paper investigates the origin of this plasticity using atomistic modeling with a recently developed machine-learning interatomic potential of -Fe. The computational results identified the precursor of crack tip plasticity, i.e. the group of activated atoms dynamically nucleated by fast crack propagation.
Tsuru, Tomohito; Han, S.*; Matsuura, Shutaro*; Chen, Z.*; Kishida, Kyosuke; Lobzenko, I.; Rao, S.*; Woodward, C.*; George, E.*; Inui, Haruyuki*
Nature Communications (Internet), 15, p.1706_1 - 1706_10, 2024/02
Times Cited Count:0Refractory high-entropy alloys (RHEAs) have attracted attention because of their potential for use in ultrahigh-temperature applications. Unfortunately, their body-centered-cubic (BCC) crystal structures make them more brittle than the ductile and fracture-resistant face-centered-cubic (FCC) HEAs. RHEAs also display significantly lower creep strengths than a leading Ni-base superalloy and its FCC matrix. To overcome these drawbacks and develop RHEAs into viable structural materials, improved fundamental understanding is needed of factors that control strength and ductility. Here we investigate two model RHEAs, TiZrHfNbTa and VNbMoTaW, and show that the former is plastically compressible down to 77 K, whereas the latter is not below 298 K. We find that hexagonal close-packed (HCP) elements in TiZrHfNbTa lower its dislocation core energy, increase its lattice distortion, and lower its shear modulus relative to VNbMoTaW whose elements are all BCC, leading to the formers higher ductility and modulus-normalized yield strength. Consistent with our yield strength models, primarily screw dislocations are present in TiZrHfNbTa after deformation, but equal numbers of edge and screw segments in VNbTaMoW. Dislocation cores are compact in VNbTaMoW and extended in TiZrHfNbTa, and different macroscopic slip planes are activated in the two RHEAs, which we attribute to the concentration of HCP elements. Our findings demonstrate how electronic structure changes related to the ratio of HCP to BCC elements can be used to control strength, ductility, and slip behavior to develop the next generation of high-temperature materials for more efficient power plants and transportation.
Suzudo, Tomoaki; Ebihara, Kenichi; Tsuru, Tomohito; Mori, Hideki*
Zairyo, 73(2), p.129 - 135, 2024/02
Body-centered-cubic transition metals, such as Fe and W, cleave along the {100} plane. To find out the mechanism of this response, atomistic simulations of curved crack-fronts of bcc Fe were conducted at 0 K using an interatomic potential created by an artificial neural network (ANN) technique. We discovered that dislocations can be emitted from the curved crack fronts along the {110} crack plane, and this phenomenon explains why the cleavage is observed only along the {100} plane. In addition, the cleavage simulations along {100} at the elevated temperature were found to be accompanied by plasticity; namely, they represented more realistic fracture.
Tsuru, Tomohito; Lobzenko, I.; Ogata, Shigenobu*; Han, W.-Z.*
Journal of Materials Research and Technology, 28, p.1013 - 1021, 2024/01
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)Some solute atoms induce hardening and embrittlement in body-centered-cubic refractory metals. Especially interstitial oxygen has a dramatic hardening effect in Nb, where the yield stress of oxygen-doped Nb alloys becomes more than twice as high as that of pure Nb. Conventional mechanisms cannot explain the oxygen-induced dramatic hardening since the interaction between dislocation and oxygen is relatively weak. Here, we focused on the three-body interaction of a screw dislocation with oxygen and vacancy. Our first-principles calculations revealed that the formation of vacancy-oxygen pair enhances the attractive interaction with a screw dislocation though the interaction between oxygen and dislocation is repulsive. Furthermore, this feature was found to be a unique nature of oxygen in Nb. The vacancy-oxygen pair increases the energy barrier for dislocation motion more significantly than an isolated vacancy and oxygen interstitial. We have discovered a new oxygen-induced mechanism: a unique octahedral-tetrahedral shuffling process of oxygen dominantly contributes to the dramatic hardening. Thus, the widely distributed vacancy-oxygen pairs behave as strong obstacles for dislocation motion that causes damage accumulation and successive hardening in oxygen-doped BCC alloys.
Yamaguchi, Masatake; Ebihara, Kenichi; Tsuru, Tomohito; Itakura, Mitsuhiro
Materials Transactions, 64(11), p.2553 - 2559, 2023/11
Times Cited Count:1 Percentile:0(Materials Science, Multidisciplinary)We attempted to calculate the hydrogen trapping energies on the incoherent interfaces of MgZn precipitates and MgSi crystallites in aluminum alloys from first-principles calculations. Since the unit cell containing the incoherent interface does not satisfy the periodic boundary condition, resulting in a discontinuity of crystal blocks, the hydrogen trapping energy was calculated in a region far from the discontinuity (vacuum) region. We found considerable trapping energies for hydrogen atoms at the incoherent interfaces consisting of assumed atomistic arrangement. We also conducted preliminary calculations of the reduction in the cohesive energy by hydrogen trapping on the incoherent interfaces of MgSi in the aluminum matrix.
precipitates and MgSi crystallites in aluminum alloys from first-principles calculations. Since the unit cell containing the incoherent interface does not satisfy the periodic boundary condition, resulting in a discontinuity of crystal blocks, the hydrogen trapping energy was calculated in a region far from the discontinuity (vacuum) region. We found considerable trapping energies for hydrogen atoms at the incoherent interfaces consisting of assumed atomistic arrangement. We also conducted preliminary calculations of the reduction in the cohesive energy by hydrogen trapping on the incoherent interfaces of MgSi in the aluminum matrix.
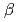 titanium alloys strong and ductile
titanium alloys strong and ductileChong, Y.*; Gholizadeh, R.*; Guo, B.*; Tsuru, Tomohito; Zhao, G.*; Yoshida, Shuhei*; Mitsuhara, Masatoshi*; Godfrey, A.*; Tsuji, Nobuhiro*
Acta Materialia, 257, p.119165_1 - 119165_14, 2023/09
Times Cited Count:4 Percentile:84.87(Materials Science, Multidisciplinary)Metastable titanium alloys possess excellent strain-hardening capability, but suffer from a low yield strength. As a result, numerous attempts have been made to strengthen this important structural material in the last decade. Here, we explore the contributions of grain refinement and interstitial additions in raising the yield strength of a Ti-12Mo (wt.%) metastable titanium alloy. Surprisingly, rather than strengthening the material, grain refinement actually lowers the ultimate tensile strength in this alloy. This unexpected and anomalous behavior is attributed to a significant enhancement in strain-induced 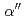 martensite phase transformation, where in-situ synchrotron X-ray diffraction analysis reveals, for the first time, that this phase is much softer than the parent phase. Instead, a combination of both oxygen addition and grain refinement is found to realize an unprecedented strength-ductility synergy in a Ti-12Mo-0.3O (wt.%) alloy. The advantageous effect of oxygen solutes in this ternary alloy is twofold. Firstly, solute oxygen largely suppresses strain-induced transformation to the martensite phase, even in a fine-grained microstructure, thus avoiding the softening effect of excessive amounts of martensite. Secondly, oxygen solutes readily segregate to twin boundaries, as revealed by atom probe tomography. This restricts the growth of
martensite phase transformation, where in-situ synchrotron X-ray diffraction analysis reveals, for the first time, that this phase is much softer than the parent phase. Instead, a combination of both oxygen addition and grain refinement is found to realize an unprecedented strength-ductility synergy in a Ti-12Mo-0.3O (wt.%) alloy. The advantageous effect of oxygen solutes in this ternary alloy is twofold. Firstly, solute oxygen largely suppresses strain-induced transformation to the martensite phase, even in a fine-grained microstructure, thus avoiding the softening effect of excessive amounts of martensite. Secondly, oxygen solutes readily segregate to twin boundaries, as revealed by atom probe tomography. This restricts the growth of 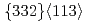 deformation twins, thereby promoting more extensive twin nucleation, leading to enhanced microstructural refinement. The insights from our work provide a cost-effective rationale for the design of strong yet tough metastable titanium alloys, with significant implications for more widespread use of this high strength-to-weight structural material.
deformation twins, thereby promoting more extensive twin nucleation, leading to enhanced microstructural refinement. The insights from our work provide a cost-effective rationale for the design of strong yet tough metastable titanium alloys, with significant implications for more widespread use of this high strength-to-weight structural material.
Lobzenko, I.; Tsuru, Tomohito; Shiihara, Yoshinori*; Iwashita, Takuya*
Materials Research Express (Internet), 10(8), p.085201_1 - 085201_12, 2023/08
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)Unlike alloys with a crystal lattice, metallic glasses (MG) do not possess distinctive defects but demonstrate a highly heterogeneous response to mechanical deformation, even in near-elastic regimes. The difficulties in describing such non-uniform behavior hamper the prediction of the mechanical properties of MGs. We apply first-principles calculations of atomic stress in CuZr MG to reveal its response to shear strain. That approach allows one to probe such parameters as displacement vector, charge transfer, or change in chemical bonds on the lowest atomic level. We find correlations between the mentioned parameters and show the importance of atomic von Mises stress in the comprehensive description of the mechanical state of a glassy system.
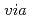 muon spin relaxation method and first-principles calculations
muon spin relaxation method and first-principles calculationsTsuru, Tomohito; Nishimura, Katsuhiko*; Matsuda, Kenji*; Nunomura, Norio*; Namiki, Takahiro*; Lee, S.*; Higemoto, Wataru; Matsuzaki, Teiichiro*; Yamaguchi, Masatake; Ebihara, Kenichi; et al.
Metallurgical and Materials Transactions A, 54(6), p.2374 - 2383, 2023/06
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)Although hydrogen embrittlement susceptibility of high-strength Al alloys is recognized as a critical issue in the practical use of Al alloys, identifying the hydrogen trapping or distribution has been challenging. In the present study, an effective approach based on experiment and simulation is proposed to explore the potential trap sites in Al alloys. Zero-field muon spin relaxation experiments were carried out for Al-0.5%Mg, Al-0.2%Cu, Al-0.15%Ti, Al-0.011%Ti, Al-0.28%V, and Al-0.015%V (at.%) in the temperature range from 5 to 300 K. The temperature variations of the dipole field widths have revealed three peaks for Al-0.5%Mg, four peaks for Al-0.2%Cu, three peaks for Al-0.011%Ti and Al-0.015%V. Atomic configurations of the muon trapping sites corresponding to the observed 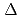 peaks are well assigned using the first-principles calculations for the trap energies of hydrogen around a solute and solute-vacancy pair. The extracted linear relationship between the muon peak temperature and the trap energy enables us to explore the potential alloying elements and their complex that have strong binding energies with hydrogen in Al alloys.
peaks are well assigned using the first-principles calculations for the trap energies of hydrogen around a solute and solute-vacancy pair. The extracted linear relationship between the muon peak temperature and the trap energy enables us to explore the potential alloying elements and their complex that have strong binding energies with hydrogen in Al alloys.
Mori, Hideki*; Tsuru, Tomohito; Okumura, Masahiko; Matsunaka, Daisuke*; Shiihara, Yoshinori*; Itakura, Mitsuhiro
Physical Review Materials (Internet), 7(6), p.063605_1 - 063605_8, 2023/06
Times Cited Count:0 Percentile:0(Materials Science, Multidisciplinary)The introduction of obstacles (e.g., precipitates) for controlling dislocation motion in molecular structures is a prevalent method for designing the mechanical strength of metals. Owing to the nanoscale size of the dislocation core (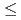 1 nm), atomic modeling is required to investigate the interactions between the dislocation and obstacles. However, conventional empirical potentials are not adequately accurate, in contrast to the calculations based on density functional theory (DFT). Therefore, the atomic-level details of the interactions between the dislocations and obstacles remain unclarified. To this end, this study applied an artificial neural network (ANN) framework to construct an atomic potential by leveraging the high accuracy of DFT. Using the constructed ANN potential, we investigated the dynamic interaction between the
1 nm), atomic modeling is required to investigate the interactions between the dislocation and obstacles. However, conventional empirical potentials are not adequately accurate, in contrast to the calculations based on density functional theory (DFT). Therefore, the atomic-level details of the interactions between the dislocations and obstacles remain unclarified. To this end, this study applied an artificial neural network (ANN) framework to construct an atomic potential by leveraging the high accuracy of DFT. Using the constructed ANN potential, we investigated the dynamic interaction between the 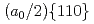 edge dislocation and obstacles in BCC iron. When the dislocation crossed the void, an ultrasmooth and symmetric half-loop was observed for the bowing-out dislocation. Except for the screw dislocation, the Peierls stress of all the dislocations predicted using the ANN was less than 100 MPa. More importantly, the results confirmed the formation of an Orowan loop in the interaction between a rigid sphere and dislocation. Furthermore, we discovered a phenomenon in which the Orowan loop disintegrated into two small loops during its interaction with the rigid sphere and dislocation.
edge dislocation and obstacles in BCC iron. When the dislocation crossed the void, an ultrasmooth and symmetric half-loop was observed for the bowing-out dislocation. Except for the screw dislocation, the Peierls stress of all the dislocations predicted using the ANN was less than 100 MPa. More importantly, the results confirmed the formation of an Orowan loop in the interaction between a rigid sphere and dislocation. Furthermore, we discovered a phenomenon in which the Orowan loop disintegrated into two small loops during its interaction with the rigid sphere and dislocation.
Lobzenko, I.; Wei, D.*; Itakura, Mitsuhiro; Shiihara, Yoshinori*; Tsuru, Tomohito
Results in Materials (Internet), 17, p.100364_1 - 100364_7, 2023/03
High-entropy alloys (HEAs) have received attention for their excellent mechanical and thermodynamic properties. A recent study revealed that Co-free face-centered cubic HEAs carried a potential to improve strength and ductility, which is of high importance for nuclear materials. Here, we implemented first-principles calculations to explore the fundamental mechanism of improving mechanical properties in Co-free HEA. We found that the local lattice distortion of Co-free HEA is more significant than that of the well-known Cantor alloy. In addition, the short-range order formation in Co-free HEA caused highly fluctuated stacking fault energy. Thus, the significant local lattice distortion and the non-uniform solid solution states composed of low- and high-stacking fault regions contribute to improving strength and ductility.
Chong, Y.*; Gholizadeh, R.*; Tsuru, Tomohito; Zhang, R.*; Inoue, Koji*; Gao, W.*; Godfrey, A.*; Mitsuhara, Masatoshi*; Morris, J. W. Jr.*; Minor, A. M.*; et al.
Nature Communications (Internet), 14, p.404_1 - 404_11, 2023/02
Times Cited Count:6 Percentile:93.59(Multidisciplinary Sciences)Interstitial oxygen embrittles titanium, particularly at cryogenic temperatures, which necessitates a stringent control of oxygen content in fabricating titanium and its alloys. Here, we propose a structural strategy, via grain refinement, to alleviate this problem. Compared to a coarse-grained counterpart that is extremely brittle at 77K, the uniform elongation of an ultrafine-grained (UFG) microstructure (grain size  2.0
2.0 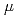 m) in Ti-0.3wt.%O was successfully increased by an order of magnitude, maintaining an ultrahigh yield strength inherent to the UFG microstructure. This unique strength-ductility synergy in UFG Ti-0.3wt.%O was achieved via the combined effects of diluted grain boundary segregation of oxygen that helps to improve the grain boundary cohesive energy and enhanced
m) in Ti-0.3wt.%O was successfully increased by an order of magnitude, maintaining an ultrahigh yield strength inherent to the UFG microstructure. This unique strength-ductility synergy in UFG Ti-0.3wt.%O was achieved via the combined effects of diluted grain boundary segregation of oxygen that helps to improve the grain boundary cohesive energy and enhanced 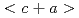 dislocation activities that contribute to the excellent strain hardening ability. The present strategy could not only boost the potential applications of high strength Ti-O alloys at low temperatures, but could also be applied to other alloy systems, where interstitial solution hardening results into an undesirable loss of ductility.
dislocation activities that contribute to the excellent strain hardening ability. The present strategy could not only boost the potential applications of high strength Ti-O alloys at low temperatures, but could also be applied to other alloy systems, where interstitial solution hardening results into an undesirable loss of ductility.
Wei, D.*; Gong, W.; Tsuru, Tomohito; Lobzenko, I.; Li, X.*; Harjo, S.; Kawasaki, Takuro; Do, H.-S.*; Bae, J. W.*; Wagner, C.*; et al.
International Journal of Plasticity, 159, p.103443_1 - 103443_18, 2022/12
Times Cited Count:27 Percentile:98.49(Engineering, Mechanical)Chong, Y.*; Tsuru, Tomohito; Guo, B.*; Gholizadeh, R.*; Inoue, Koji*; Tsuji, Nobuhiro*
Acta Materialia, 240, p.118356_1 - 118356_15, 2022/11
Times Cited Count:15 Percentile:92.67(Materials Science, Multidisciplinary)In this study, we systematically investigated the influences of nitrogen content and grain size on the tensile properties and deformation behaviors of titanium at room temperature. By high-pressure torsion and annealing, we obtained ultrafine-grained (UFG) Ti-0.3wt.%N alloy with a fully recrystallized microstructure, which combined an unprecedented synergy of ultrahigh yield strength (1.04 GPa) and large uniform elongation (10%). The hardening and strain-hardening mechanisms of Ti-0.3wt.%N alloy were comprehensively studied via deformation substructure observation and first-principles calculations. It is revealed that the contributions of nitrogen to the excellent strength/ductility balance in UFG Ti-0.3wt.%N were twofold. On one hand, nitrogen atoms inside the grains strongly impeded the motion of 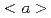 dislocations on prismatic plane due the shuffling of nitrogen from octahedral to hexahedral site, giving rise to a six-fold increase in the friction stress than pure Ti. Moreover, the greatly reduced stacking fault energy difference between prismatic and pyramidal planes in Ti-0.3wt.%N alloy facilitated an easier activation of dislocations, which contributed to an enhanced strain-hardening rate. On the other hand, some nitrogen atoms segregated near the grain boundaries, a phenomenon discovered in -titanium for the first time. These segregated nitrogen atoms served as an additional contributor to the yield strength of UFG Ti-0.3wt.%N, by raising the barrier against dislocation slip transfer between grains. Our experimental and theoretical calculation work provide insights for the design of affordable high strength titanium without a large sacrifice of ductility, shedding lights on a more widespread use of this high strength to weight material.
dislocations on prismatic plane due the shuffling of nitrogen from octahedral to hexahedral site, giving rise to a six-fold increase in the friction stress than pure Ti. Moreover, the greatly reduced stacking fault energy difference between prismatic and pyramidal planes in Ti-0.3wt.%N alloy facilitated an easier activation of dislocations, which contributed to an enhanced strain-hardening rate. On the other hand, some nitrogen atoms segregated near the grain boundaries, a phenomenon discovered in -titanium for the first time. These segregated nitrogen atoms served as an additional contributor to the yield strength of UFG Ti-0.3wt.%N, by raising the barrier against dislocation slip transfer between grains. Our experimental and theoretical calculation work provide insights for the design of affordable high strength titanium without a large sacrifice of ductility, shedding lights on a more widespread use of this high strength to weight material.
 neutron diffraction
neutron diffractionWei, D.*; Gong, W.; Tsuru, Tomohito; Kawasaki, Takuro; Harjo, S.; Cai, B.*; Liaw, P. K.*; Kato, Hidemi*
International Journal of Plasticity, 158, p.103417_1 - 103417_17, 2022/11
Times Cited Count:22 Percentile:97.48(Engineering, Mechanical)Suzudo, Tomoaki; Ebihara, Kenichi; Tsuru, Tomohito; Mori, Hideki*
Scientific Reports (Internet), 12, p.19701_1 - 19701_10, 2022/11
Times Cited Count:4 Percentile:41.53(Multidisciplinary Sciences)Body-centered-cubic (bcc) transition metals, such as -Fe and W, cleave along the {100} plane, even though the surface energy is the lowest along the {110} plane. To unravel the mechanism of this odd response, large-scale atomistic simulations of curved cleavage cracks of -Fe were conducted in association with stress intensity factor analyses of straight crack fronts using an interatomic potential created by an artificial neural network technique. The study provides novel findings: Dislocations are emitted from the crack fronts along the {110} cleavage plane, and this phenomenon explains why the {100} plane can be the cleavage plane. However, the simple straight crack-front analyses did not yield the same conclusion. It is suggested that atomistic modeling, at sufficiently large scales to capture the inherent complexities of materials using highly accurate potentials, is necessary to correctly predict the mechanical strength. The method adopted in this study is generally applicable to the cleavage problem of bcc transition metals and alloys.
Tsuru, Tomohito
Zairyo, 71(8), p.660 - 665, 2022/08
The dynamic behavior of individual defects at the nanoscale plays an important role in understanding the mechanical properties of highly controlled materials and the nature of their mechanical functions. The purpose of this study is to reveal the origin of the mechanical properties from the electronic structure calculations of dislocation core. In this paper, we propose a modeling that describes the slip bahavior based on the kink mechanism for alloys with a body-centered cubic lattice structure (BCC) that shows a unusual temperature dependence on mechanical properties. In addition, we introduce analytical model to understand the role of alloying elements on dislocation motion from the electronic structure and predict mechanical properties.
Kurihara, Kensuke*; Lobzenko, I.; Tsuru, Tomohito; Serizawa, Ai*
Keikinzoku, 72(7), p.427 - 429, 2022/07
Nanoclusters formed of the Al-Mg-Si alloy affect the aging behavior of the alloy depending on the formation temperature. Since Al, Mg and Si have adjacent atomic numbers, it is difficult to analyze them using the X-ray diffraction method. Therefore, in recent years, Al-Mg-Ge alloys in which Si is replaced with the homologous element Ge have been used. Attempts have been made to analyze the structure of the precipitate. In this study, we quantitatively evaluate the interaction between solute atoms and pores in Al-Mg-Si alloys and Al-Mg-Ge alloys using first-principles calculations based on the density general function theory, and solute atoms. From the viewpoint of bond stability between pores and pores, the precipitation behavior of both alloys was compared and examined.
Yamaguchi, Masatake; Tsuru, Tomohito; Itakura, Mitsuhiro; Abe, Eiji*
Scientific Reports (Internet), 12(1), p.10886_1 - 10886_7, 2022/07
Times Cited Count:0 Percentile:0(Multidisciplinary Sciences)no abstracts in English
Tsuru, Tomohito; Lobzenko, I.; Wei, D.*
Modelling and Simulation in Materials Science and Engineering, 30(2), p.024003_1 - 024003_11, 2022/03
Times Cited Count:8 Percentile:77.62(Materials Science, Multidisciplinary)High-entropy alloys (HEA) have been receiving increased attention for their excellent mechanical properties. Our recent study revealed that Si-doped face-centered cubic (FCC) HEAs have great potential to improve both strength and ductility. Here, we carried out first-principles calculations in cooperation with Monte Carlo simulation and structural factor analysis to explore the effect of Si addition on the macroscopic mechanical properties. As a result, Si addition increased the local lattice distortion and the stacking fault energy. Furthermore, the SRO formation in Si-doped alloy caused highly fluctuated SF energy. Thus, the heterogeneous solid solution states in which low and high SF regions are distributed into the matrix were nucleated. This unique feature in Si-doped FCC-HEA induces ultrafine twin formation in Si-doped alloys, which can be a dominant factor in improving both strength and ductility.
Wei, D.*; Wang, L.*; Zhang, Y.*; Gong, W.; Tsuru, Tomohito; Lobzenko, I.; Jiang, J.*; Harjo, S.; Kawasaki, Takuro; Bae, J. W.*; et al.
Acta Materialia, 225, p.117571_1 - 117571_16, 2022/02
Times Cited Count:59 Percentile:99.75(Materials Science, Multidisciplinary)