


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 -iron
-ironYoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Kimizuka, Hajime*; Shiga, Motoyuki
Journal of Physical Chemistry C, 116(43), p.23113 - 23119, 2012/11
Times Cited Count:18 Percentile:47.11(Chemistry, Physical)The diffusion coefficients of hydrogen (H) and tritium (T) in -Fe have been computed using two approximate quantum dynamical techniques, i.e. centroid molecular dynamics (CMD) and ring polymer molecular dynamics (RPMD), in the temperature range of T = 100-1000 K using the embedded-atom-method (EAM) potential. It has been found that the RPMD and CMD methods give very similar results. From a further analysis based on quantum transition state theory (centroid density QTST) combined with path integral molecular dynamics (PIMD), it has been clear that there is a crossover between thermal and quantum mechanisms at about T = 500 K and 300 K for H and T diffusions, respectively. The importance of nuclear quantum effects at low temperatures has been illustrated in terms of the effective free energy surface map.
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics, 394(1), p.46 - 51, 2012/02
Times Cited Count:18 Percentile:53.54(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed for porphycene and its isotopic variants in order to understand the effect of isotopic substitution of inner protons on the double proton transfer mechanism. Our quantum simulations show that double proton transfer of the unsubstituted porphycene at the room temperature mainly occurs via a so-called concerted mechanism through the second-order saddle point. In addition, we found that both isotopic substitution and temperature significantly affect the double proton transfer mechanism. For example, the contribution of the stepwise mechanism increases with a temperature increase. It has been found that out-of-plane vibrational motions significantly decrease the contribution of the concerted proton transfer mechanism.
Sugawara, Shuichi*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Physical Chemistry A, 115(42), p.11486 - 11494, 2011/09
Times Cited Count:20 Percentile:56.15(Chemistry, Physical)We have carried out path integral molecular dynamics simulations for hydrated sulfuric acid clusters to understand acid-dissociation and hydrogen-bonded structural rearrangement processes in these clusters from a quantum mechanical viewpoint. We have found that the acid dissociation processes, first and second deprotonation, effectively occur in a hydrated cluster with a specific cluster size. The mechanisms of the proton-transfer processes were analyzed in detail and it was found that the distance between O in sulfuric acid and O in the proton-accepting water is playing an important role. We also found that the water coordination number of the proton-accepting water is important in the proton-transfer processes.
Yoshikawa, Takehiro*; Sugawara, Shuichi*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Chemical Physics Letters, 496(1-3), p.14 - 19, 2010/08
Times Cited Count:28 Percentile:67.01(Chemistry, Physical)Full-dimensional path-integral molecular dynamics simulations were performed to determine whether the double proton transfer tautomerization of porphycene is a concerted or a stepwise process. It was found that double proton transfer occurs dominantly through the concerted pathway via the second-order saddle point structure and that contribution of the stepwise mechanism increases with a temperature increase. Nuclear quantum effects play essential roles in determining the proton transfer mechanism.
 O)
O)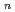 (
(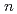 = 1-7) clusters on semi-empirical PM6 potential energy surfaces
= 1-7) clusters on semi-empirical PM6 potential energy surfacesYoshikawa, Takehiro*; Motegi, Haruki*; Kakizaki, Akira*; Takayanagi, Toshiyuki*; Shiga, Motoyuki
Chemical Physics, 365(1-2), p.60 - 68, 2009/12
Times Cited Count:15 Percentile:44.84(Chemistry, Physical)Path-integral molecular dynamics simulations for the hydrogen-bonded glycine-(HO) ( = 1-7) clusters have been carried out using an on-the-fly direct dynamics technique at the semiempiricalPM6 level of theory. In the case of smaller clusters with = 1-3, the simulations show that the clusterstructure takes exclusively the hydrogen-bonded complex between a canonical neutral glycine and a water cluster moiety. In contrast, it was found that proton-exchange processes effectively occur between the COOH carboxylic group and water cluster moiety for = 4-6 clusters although the overall structures are the complex between a neutral glycine and water clusters. In the case of the = 7 cluster, glycine preferentially takes a zwitterionic form having NH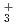 and COO
and COO functional groups.
functional groups.
O)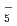 and (DO)
and (DO)Takayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Motegi, Haruki*; Shiga, Motoyuki
Chemical Physics Letters, 482(4-6), p.195 - 200, 2009/12
Times Cited Count:10 Percentile:28.06(Chemistry, Physical)Path-integral molecular dynamics simulations have been performed for water anion clusters,(HO) and (DO), on the basis of a semiempirical one-electron pseudopotential polarization model. Due to larger zero-point vibrational amplitudes for H atoms than that of D atoms, hydrogen-bond lengths in the (HO) cluster are slightly larger than those in (DO). The distribution of the vertical detachment energies for (HO) also show a broader feature than that for (DO). The present simulations thus demonstrate the importance of nuclear quantum effects in water anion clusters.
SO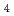
 (HO) (=1-6) on semiempirical PM6 potential surfaces
(HO) (=1-6) on semiempirical PM6 potential surfacesKakizaki, Akira*; Motegi, Haruki*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
Quantum path-integral molecular dynamics simulations for a series of sulfuric acid clusters, HSO(HO)(=1-6), were performed using semiempirical PM6 method. It was found that the acid dissociation probability increases with an increase in the cluster size, and that so-called contact-ion-pair structures are dominant in the proton-dissociated clusters. The importance of nuclear quantum effects in the cluster structures and proton-transfer processes is demonstrated.
 O)
O)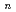 (=2-7) clusters on semiempirical PM6 potential energy surfaces
(=2-7) clusters on semiempirical PM6 potential energy surfacesTakayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
Molecular dynamics simulations based on semiempirical PM6 method was carried out to study statistical structures of glycine-water clusters. We found that zwitterionic form is more stable than neutral form as the cluster size increases. The difference in water solvation structures around the neutral or zwitterionic glycine form is also discussed.
