


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Li, F.*; Tang, X.*; Fei, Y.*; Zhang, J.*; Liu, J.*; Lang, P.*; Che, G.*; Zhao, Z.*; Zheng, Y.*; Fang, Y.*; et al.
Journal of the American Chemical Society, 147(17), p.14054 - 14059, 2025/04
Times Cited Count:1 Percentile:0.00(Chemistry, Multidisciplinary)We synthesized a crystalline graphane nanoribbon (GANR) via pressure-induced polymerization of 2,2'-bipyrazine (BPZ). By performing Rietveld refinement of in situ neutron diffraction data, nuclear magnetic resonance spectroscopy, infrared spectra, and theoretical calculation, we found that BPZ experienced Diels-Alder polymerization between the 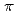
 stacked aromatic rings, and formed extended boat-GANR structures with exceptional long-range order. The unreacted -C=N- groups bridge the two ends of the boat, and ready for further functionalization. The GANR has a bandgap of 2.25 eV, with booming photoelectric response (
stacked aromatic rings, and formed extended boat-GANR structures with exceptional long-range order. The unreacted -C=N- groups bridge the two ends of the boat, and ready for further functionalization. The GANR has a bandgap of 2.25 eV, with booming photoelectric response (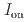 /
/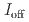 =18.8). Our work highlights that the high-pressure topochemical polymerization is a promising method for the precise synthesis of graphane with specific structure and desired properties.
=18.8). Our work highlights that the high-pressure topochemical polymerization is a promising method for the precise synthesis of graphane with specific structure and desired properties.
Zhang, X.-T.*; Xing, Y.-H.*; Yao, X.-P.*; Ominato, Yuya*; Zhang, L.*; Matsuo, Mamoru
Communications Physics (Internet), 8, p.103_1 - 103_8, 2025/03
Times Cited Count:0 Percentile:0.00(Physics, Multidisciplinary)Rajeev, H. S.*; Hu, X.*; Chen, W.-L.*; Zhang, D.*; Chen, T.*; Kofu, Maiko*; Kajimoto, Ryoichi; Nakamura, Mitsutaka; Chen, A. Z.*; Johnson, G. C.*; et al.
Journal of the Physical Society of Japan, 94(3), p.034602_1 - 034602_14, 2025/03
Times Cited Count:0 Percentile:0.00(Physics, Multidisciplinary)Brumm, S.*; Gabrielli, F.*; Sanchez Espinoza, V.*; Stakhanova, A.*; Groudev, P.*; Petrova, P.*; Vryashkova, P.*; Ou, P.*; Zhang, W.*; Malkhasyan, A.*; et al.
Annals of Nuclear Energy, 211, p.110962_1 - 110962_16, 2025/02
Times Cited Count:6 Percentile:94.85(Nuclear Science & Technology) neutron imaging and diffraction analysis revealing spatial lithiation phase evolution in an ultra-thick graphite electrode
neutron imaging and diffraction analysis revealing spatial lithiation phase evolution in an ultra-thick graphite electrodeStrobl, M.*; Baur, M. E.*; Samothrakitis, S.*; Molamud, F.*; Zhang, X.*; Tung, P. K. M.*; Schmidt, S.*; Woracek, R.*; Lee, J.*; Kiyanagi, Ryoji; et al.
Advanced Energy Materials, p.2405238_1 - 2405238_9, 2025/01
Times Cited Count:0Yang, X.*; Che, G.*; Wang, Y.*; Zhang, P.*; Tang, X.*; Lang, P.*; Gao, D.*; Wang, X.*; Wang, Y.*; Hattori, Takanori; et al.
Nano Letters, 25(3), p.1028 - 1035, 2025/01
Times Cited Count:1 Percentile:84.76(Chemistry, Multidisciplinary)Saturated sp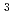 -carbon nanothreads (CNTh) have garnered significant interest due to their predicted high Young's modulus and thermal conductivity. While the incorporation of heteroatoms into the central ring has been shown to influence the formation of CNTh and yield chemically homogeneous products, the impact of pendant groups on the polymerization process remains underexplored. In this study, we investigate the pressure-induced polymerization of phenol, revealing two phase transitions occurring below 0.5 and 4 GPa. Above 20 GPa, phenol polymerizes into degree-4 CNThs featuring hydroxyl and carbonyl groups. Hydrogen transfer of hydroxyl groups was found to hinder the formation of degree-6 nanothreads. Our findings highlight the crucial role of the hydroxyl group in halting further intracolumn polymerization and offer valuable insights for future mechanism research and nanomaterial synthesis.
-carbon nanothreads (CNTh) have garnered significant interest due to their predicted high Young's modulus and thermal conductivity. While the incorporation of heteroatoms into the central ring has been shown to influence the formation of CNTh and yield chemically homogeneous products, the impact of pendant groups on the polymerization process remains underexplored. In this study, we investigate the pressure-induced polymerization of phenol, revealing two phase transitions occurring below 0.5 and 4 GPa. Above 20 GPa, phenol polymerizes into degree-4 CNThs featuring hydroxyl and carbonyl groups. Hydrogen transfer of hydroxyl groups was found to hinder the formation of degree-6 nanothreads. Our findings highlight the crucial role of the hydroxyl group in halting further intracolumn polymerization and offer valuable insights for future mechanism research and nanomaterial synthesis.
Xu, J.*; Lang, P.*; Liang, S.*; Zhang, J.*; Fei, Y.*; Wang, Y.*; Gao, D.*; Hattori, Takanori; Abe, Jun*; Dong, X.*; et al.
Journal of Physical Chemistry Letters (Internet), p.2445 - 2451, 2025/00
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)The Alder-ene reaction is a chemical reaction between an alkene with an allylic hydrogen, and it provides an efficient method to construct the C-C bond. Traditionally, this reaction requires catalysts, high temperatures, or photocatalysis. In this study, we reported a high-pressure-induced solid-state Alder-ene reaction of 1-hexene at room temperature without a catalyst. 1-Hexene crystallizes at 4.3 GPa and polymerizes at 18 GPa, forming olefins. By exploring gas chromatography-mass spectrometry, we discovered that 1-hexene generates dimeric products through the Alder-ene reaction under high pressures. The in situ neutron diffraction shows that the reaction process did not obey the topochemical rule. A six-membered ring transition state including one C-H 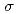 bond and two alkene bonds was evidenced by the theoretical calculation, whose energy obviously decreased when compressed to 20 GPa. Our work offers a novel and promising method to realize the Alder-ene reaction at room temperature without a catalyst, expanding the application of this important reaction.
bond and two alkene bonds was evidenced by the theoretical calculation, whose energy obviously decreased when compressed to 20 GPa. Our work offers a novel and promising method to realize the Alder-ene reaction at room temperature without a catalyst, expanding the application of this important reaction.
Liu, P.-F.*; Li, X.*; Li, J.*; Zhu, J.*; Tong, Z.*; Kofu, Maiko*; Nirei, Masami; Xu, J.*; Yin, W.*; Wang, F.*; et al.
National Science Review, 11(12), p.nwae216_1 - nwae216_10, 2024/12
Times Cited Count:13 Percentile:94.32(Multidisciplinary Sciences)Zhang, Y.-J.*; Umeda, Takemasa*; Morooka, Satoshi; Harjo, S.; Miyamoto, Goro*; Furuhara, Tadashi*
Metallurgical and Materials Transactions A, 55(10), p.3921 - 3936, 2024/10
Times Cited Count:0 Percentile:0.00(Materials Science, Multidisciplinary)Ying, H.*; Yang, X.*; He, H.*; Yan, A.*; An, K.*; Ke, Y.*; Wu, Z.*; Tang, S.*; Zhang, Z.*; Dong, H.*; et al.
Scripta Materialia, 250, p.116181_1 - 116181_7, 2024/09
Times Cited Count:2 Percentile:36.18(Nanoscience & Nanotechnology)Qin, T. Y.*; Hu, F. F.*; Xu, P. G.; Zhang, H.*; Zhou, L.*; Ao, N.*; Su, Y. H.; Shobu, Takahisa; Wu, S. C.*
International Journal of Fatigue, 185, p.108336_1 - 108336_13, 2024/08
Times Cited Count:9 Percentile:93.27(Engineering, Mechanical)Watanabe, Miku*; Miyamoto, Goro*; Zhang, Y.*; Morooka, Satoshi; Harjo, S.; Kobayashi, Yasuhiro*; Furuhara, Tadashi*
ISIJ International, 64(9), p.1464 - 1476, 2024/07
Times Cited Count:3 Percentile:57.76(Metallurgy & Metallurgical Engineering)Zhou, L.*; Zhang, H.*; Qin, T. Y.*; Hu, F. F.*; Xu, P. G.; Ao, N.*; Su, Y. H.; He, L. H.*; Li, X. H.*; Zhang, J. R.*; et al.
Metallurgical and Materials Transactions A, 55(7), p.2175 - 2185, 2024/07
Times Cited Count:3 Percentile:70.74(Materials Science, Multidisciplinary)Zeng, Z.*; Zhou, C.*; Zhou, H.*; Han, L.*; Chi, R.*; Li, K.*; Kofu, Maiko; Nakajima, Kenji; Wei, Y.*; Zhang, W.*; et al.
Nature Physics, 20(7), p.1097 - 1102, 2024/07
Times Cited Count:12 Percentile:94.31(Physics, Multidisciplinary)Li, L.*; Miyamoto, Goro*; Zhang, Y.*; Li, M.*; Morooka, Satoshi; Oikawa, Katsunari*; Tomota, Yo*; Furuhara, Tadashi*
Journal of Materials Science & Technology, 184, p.221 - 234, 2024/06
Times Cited Count:7 Percentile:52.51(Materials Science, Multidisciplinary)Wang, S.*; Wang, J.*; Zhang, S.*; Wei, D.*; Chen, Y.*; Rong, X.*; Gong, W.; Harjo, S.; Liu, X.*; Jiao, Z.*; et al.
Journal of Materials Science & Technology, 185, p.245 - 258, 2024/06
Times Cited Count:17 Percentile:98.00(Materials Science, Multidisciplinary)
Liao, J.*; Huang, Z.*; Shangguan, Y.*; Zhang, B.*; Cheng, S.*; Xu, H.*; Kajimoto, Ryoichi; Kamazawa, Kazuya*; Bao, S.*; Wen, J.*
Physical Review B, 109(22), p.224411_1 - 224411_10, 2024/06
Times Cited Count:0 Percentile:0.00(Materials Science, Multidisciplinary)Baccou, J.*; Glantz, T.*; Ghione, A.*; Sargentini, L.*; Fillion, P.*; Damblin, G.*; Sueur, R.*; Iooss, B.*; Fang, J.*; Liu, J.*; et al.
Nuclear Engineering and Design, 421, p.113035_1 - 113035_16, 2024/05
Times Cited Count:6 Percentile:96.49(Nuclear Science & Technology)Guo, B.*; Chen, H.*; Chong, Y.*; Mao, W.; Harjo, S.; Gong, W.; Zhang, Z.*; Jonas, J. J.*; Tsuji, Nobuhiro*
Acta Materialia, 268, p.119780_1 - 119780_11, 2024/04
Times Cited Count:10 Percentile:94.16(Materials Science, Multidisciplinary)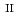 complex exhibiting intermolecular proton shifting coupled spin transition
complex exhibiting intermolecular proton shifting coupled spin transitionJi, T.*; Su, S.*; Wu, S.*; Hori, Yuta*; Shigeta, Yasuteru*; Huang, Y.*; Zheng, W.*; Xu, W.*; Zhang, X.*; Kiyanagi, Ryoji; et al.
Angewandte Chemie; International Edition, 63(25), p.e202404843_1 - e202404843_6, 2024/04
Times Cited Count:0 Percentile:0.00(Chemistry, Multidisciplinary)