


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Saeki, Morihisa; Akagi, Hiroshi; Fujii, Masaaki*
Journal of Chemical Theory and Computation, 2(4), p.1176 - 1183, 2006/07
Times Cited Count:31 Percentile:70.92(Chemistry, Physical)The structures of the naphthalene monomer and dimer were investigated by performing vibrational analysis. The optimized geometry of the naphthalene monomer was found to be distorted out of plane in MP2/6-31G, MP2/6-31G*, MP2/6-31+G*, MP2/6-311G calculations, while it maintains the plane of symmetry in MP2/6-31G*(0.25) and MP2/cc-pVDZ. Based on the result of the geometrical optimization in the monomer, the MP2/cc-pVDZ level was employed in a calculation of the dimer. The naphthalene dimer has four stable isomers: the naphthalene molecules are bound by the 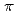 - interaction in three isomers and by the C-H... interaction in one isomer. The relative stability of the isomers suggested an enhancement of the - interaction in the structures with lower symmetry, which could be explained using a molecular-orbital model. Based on the calculated frequencies and the infrared spectrum, the vibrational modes available to experimentally distinguish the isomers were predicted.
- interaction in three isomers and by the C-H... interaction in one isomer. The relative stability of the isomers suggested an enhancement of the - interaction in the structures with lower symmetry, which could be explained using a molecular-orbital model. Based on the calculated frequencies and the infrared spectrum, the vibrational modes available to experimentally distinguish the isomers were predicted.
Matsumoto, Atsushi; Tobias, I.*; Olson, W. K.*
Journal of Chemical Theory and Computation, 1(1), p.117 - 129, 2005/01
Times Cited Count:11 Percentile:35.65(Chemistry, Physical)We have extended a newly developed approach to study the low frequency normal modes of mesoscopic fragments of linear DNA in order to investigate the dynamics of closed circular molecules of comparable size, i.e., a few hundred base pairs. We have added restraint energy terms and a global minimization step to treat the more complicated, spatially constrained duplex in terms of the intrinsic conformation and flexibility of the constituent base-pair "step" parameters.
Matsumoto, Atsushi; Tobias, I.*; Olson, W. K.*
Journal of Chemical Theory and Computation, 1(1), p.130 - 142, 2005/01
Times Cited Count:9 Percentile:30.01(Chemistry, Physical)Fine structural and energetic details embedded in the DNA base sequence, such as intrinsic curvature, are important to the packaging and processing of the genetic material. Here we investigate the internal dynamics of a 200 bp closed circular molecule with natural curvature using a newly developed normal-mode treatment of DNA in terms of neighboring base-pair "step" parameters. As superhelical stress is accumulated in the DNA, the frequency, i.e., energy, of the dominant bending mode decreases in value, and if the imposed stress is sufficiently large, a global configurational rearrangement of the circle to the figure-8 form takes place. We combine energy minimization with normal-mode calculations of the two states to decipher the configurational pathway between the two states.
