


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Kai, Takeshi; Toigawa, Tomohiro; Matsuya, Yusuke*; Hirata, Yuho; Tsuchida, Hidetsugu*; Yokoya, Akinari*
Journal of Chemical Physics, 162(15), p.154102_1 - 154102_11, 2025/04
Times Cited Count:0Scientific knowledge of low-energy electrons resulting from water radiolysis is required to estimate radiation DNA damage. However, since the analysis of water radiolysis is very complex, this study focuses on the experimental values of low-energy electrons related to simple water photolysis and those generated by photoirradiation of electrodes in water. Both experimental analyses involve the presence or absence of a Coulomb field in the parent ion. In this study, we analyzed these experimental values using a calculation code that combines Monte Carlo and molecular dynamics methods. As a result, it was shown that the code reproduced the experimental values even under different experimental conditions, and the code was validated. The calculation code will be a powerful tool for analyzing the interaction between low-energy electrons and DNA, and is expected to be applied to elucidate the formation mechanism of radiation DNA damage.
Matsuya, Yusuke; Yoshii, Yuji*; Kusumoto, Tamon*; Ogawa, Tatsuhiko; Onishi, Seiki*; Hirata, Yuho; Sato, Tatsuhiko; Kai, Takeshi
Physical Chemistry Chemical Physics, 27(14), p.6887 - 6898, 2025/04
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Radicals by water radiolysis play an important role in evaluating radiation-induced biological effects, such as DNA damage induction, chromosomal aberrations, and carcinogenesis. In the Particle and Heavy Ion Transport code System (PHITS), a track-structure simulation mode enabling the estimation of each atomic interactions in water and a chemical simulation code (PHITS-Chem) dedicated to electron beams that can simulate radical dynamics have been developed in our previous study. Here, we developed the PHITS-Chem code applicable to any ion species, considering a space partitioning method to detect radical reactions more efficiently and the 4D visualization function. The updated PHITS-Chem code was verified by comparing the simulated G values of proton beams,  particle beams, and carbon ion beams to the corresponding values in the literature. We succeeded in intuitively evaluating the diffusion dynamics of radicals using the PHITS 3D drawing software, PHIG-3D. The time to calculate the G values was reduced (e.g., about 28 times faster) while maintaining its calculation accuracy. The developed PHITS-Chem code is expected to contribute to precise and intuitive understanding of the biological effects induced by radicals in ion-beam radiotherapy.
particle beams, and carbon ion beams to the corresponding values in the literature. We succeeded in intuitively evaluating the diffusion dynamics of radicals using the PHITS 3D drawing software, PHIG-3D. The time to calculate the G values was reduced (e.g., about 28 times faster) while maintaining its calculation accuracy. The developed PHITS-Chem code is expected to contribute to precise and intuitive understanding of the biological effects induced by radicals in ion-beam radiotherapy.
Che, G.*; Fei, Y.*; Tang, X.*; Zhao, Z.*; Hattori, Takanori; Abe, Jun*; Wang, X.*; Ju, J.*; Dong, X.*; Wang, Y.*; et al.
Physical Chemistry Chemical Physics, 27(2), p.1112 - 1118, 2025/01
Times Cited Count:3 Percentile:65.57(Chemistry, Physical)Pressure-induced polymerization (PIP) of aromatic molecules has emerged as an effective method for synthesizing various carbon-based materials. In this work, PIP of 1,4-difluorobenzene (1,4-DFB) was investigated. 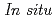 high-pressure investigations of 1,4-DFB reveal a phase transition at approximately 12.0 GPa and an irreversible chemical reaction at 18.7 GPa. Structural analysis of the product and the kinetics of the reaction uncovered the formation of pseudohexagonal stacked fluoro-diamond nanothreads with linear growth. Compared to the crystal structures of benzene under high pressure, 1,4-DFB exhibits higher compression along the [001] axis. The anisotropic compression is attributed to the stronger H
high-pressure investigations of 1,4-DFB reveal a phase transition at approximately 12.0 GPa and an irreversible chemical reaction at 18.7 GPa. Structural analysis of the product and the kinetics of the reaction uncovered the formation of pseudohexagonal stacked fluoro-diamond nanothreads with linear growth. Compared to the crystal structures of benzene under high pressure, 1,4-DFB exhibits higher compression along the [001] axis. The anisotropic compression is attributed to the stronger H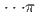 interaction along the [01
interaction along the [01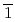 ] axis and the potential compression-inhibiting H
] axis and the potential compression-inhibiting H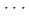 F interactions along the [100] and [010] axes, and it facilitates a possible reaction pathway along the [01] axis. This work emphasizes the crucial role of functionalization in modulating molecular stacking and influencing the reaction pathway.
F interactions along the [100] and [010] axes, and it facilitates a possible reaction pathway along the [01] axis. This work emphasizes the crucial role of functionalization in modulating molecular stacking and influencing the reaction pathway.
Sugiyama, Hitoshi*; Kato, Kenichi*; Sekine, Naoko*; Sekine, Yurina; Watanabe, Tomoaki*; Fukazawa, Tomoko*
Chemical Physics Letters, 856, p.141655_1 - 141655_8, 2024/12
Times Cited Count:1 Percentile:42.16(Chemistry, Physical)To investigate the effects of polymer hydrophilicity on structures of water in hydrogels, differential scanning calorimetry and X-ray diffraction measurements were performed. The results show that the amount of intermediate water in polyacrylamide (PAA) hydrogel is about 12% smaller than that in poly-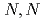 -dimethylacrylamide (PDMAA) hydrogel. Furthermore, it was found that the bound water in PAA hydrogel primarily exists around the surface of the polymer bundles, whereas that in PDMAA hydrogel acts as a crosslinker factor for dehydration and water absorption in hydrogels.
-dimethylacrylamide (PDMAA) hydrogel. Furthermore, it was found that the bound water in PAA hydrogel primarily exists around the surface of the polymer bundles, whereas that in PDMAA hydrogel acts as a crosslinker factor for dehydration and water absorption in hydrogels.
 and machine learning potentials for modeling nuclear quantum effects in water
and machine learning potentials for modeling nuclear quantum effects in waterThomsen, B.; Nagai, Yuki*; Kobayashi, Keita; Hamada, Ikutaro*; Shiga, Motoyuki
Journal of Chemical Physics, 161(20), p.204109_1 - 204109_18, 2024/11
Times Cited Count:1 Percentile:42.16(Chemistry, Physical)We introduce the self-learning path integral hybrid Monte Carlo with mixed and machine learning potentials (SL-PIHMC-MIX) method which allows the application of hybrid Monte Carlo for both path integrals and for larger system sizes. The method shows savings of an order of magnitude with respect to the number of DFT calculations needed to calculate and converge the structure of room temperature water when using SL-PIHMC-MIX over ab initio path integral molecular dynamics (PIMD).
Yoshimune, Wataru*; Higuchi, Yuki*; Song, F.; Hibi, Shogo*; Matsumoto, Yoshihiro*; Hayashida, Hirotoshi*; Nozaki, Hiroshi*; Shinohara, Takenao; Kato, Satoru*
Physical Chemistry Chemical Physics, 26(47), p.29466 - 29474, 2024/11
Times Cited Count:1 Percentile:42.16(Chemistry, Physical)Tsuchida, Hidetsugu*; Tezuka, Tomoya*; Kai, Takeshi; Matsuya, Yusuke*; Majima, Takuya*; Saito, Manabu*
Journal of Chemical Physics, 161(10), p.104503_1 - 104503_8, 2024/09
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Although fast ion beams can damage DNA by chemical products such as secondary electrons produced by their interaction with water in living cells, the process of formation of these chemical products in the Bragg peak region used in particle therapy is not fully understood. To investigate this process, we performed experiments to evaluate the yields of radiolytic products produced when a liquid water jet in vacuum is irradiated with a MeV-energy carbon beam. In addition, ionization processes in water due to incident ions and secondary electrons were simulated using a radiation transport Monte Carlo code. The results indicated that the primary source of ionization in water is secondary electrons. Finally, we show that these elementary processes contribute to the development of radiation biophysics and biochemistry to study the formation mechanism of DNA damage.
Nakata, Yuto; Sasaki, Takehiko*; Thomsen, B.; Shiga, Motoyuki
Chemical Physics Letters, 845, p.141285_1 - 141285_9, 2024/06
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Using density functional theory and metadynamics simulations, we study cellobiose hydrolysis and glucose hydrogenation with silica-supported platinum and palladium catalysts in hot water, relevant to green cellulose conversion. It is found that cellobiose hydrolysis can proceed by the attack of hydrogen atoms adsorbed on metal or protons spilled over to silica forming glucose. Glucose can then be hydrogenated by hydrogen atoms adsorbed at platinum/water interface forming sorbitol. The reaction barriers of hydrolysis and hydrogenation at platinum/water interface are both lower than that at palladium/water interface, which explains the experimental finding that the platinum performs as a better catalyst than palladium.
Toigawa, Tomohiro; Kai, Takeshi; Kumagai, Yuta; Yokoya, Akinari*
Journal of Chemical Physics, 160(21), p.214119_1 - 214119_9, 2024/06
Times Cited Count:3 Percentile:65.57(Chemistry, Physical)The spur reaction is crucial for determining radiolysis or photolysis in liquid, but the spur expansion process has yet to be elucidated. One reason is the need to understand the role of the dielectric response of the solvating molecules surrounding the charged species generated by ionization. The dielectric response corresponds to the time evolution of the permittivity and might affect the chemical reaction-diffusion of the species in a spur expansion process. This study examined the competitive relationship between reaction-diffusion kinetics and the dielectric response by solving the Debye-Smoluchowski equation while considering the dielectric response. The Coulomb force between the charged species gradually decreases with the dielectric response. Our calculation results found a condition where fast recombination occurs before the dielectric response is complete. Although it has been reported that the primary G-values of free electrons depend on the static dielectric constant under low-linear-energy transfer radiation-induced ionization, we propose that considering the dielectric response can provide a deeper insight into fast recombination reactions under high-linear-energy transfer radiation- or photo-induced ionization. Our simulation method enables the understanding of fast radiation-induced phenomena in liquids.
 He spin filter and small-angle X-ray scattering
He spin filter and small-angle X-ray scatteringRyoki, Akiyuki*; Watanabe, Fumi*; Okudaira, Takuya*; Takahashi, Shingo*; Oku, Takayuki; Hiroi, Kosuke; Motokawa, Ryuhei; Nakamura, Yo*
Journal of Chemical Physics, 160(11), p.114907_1 - 114907_9, 2024/03
Times Cited Count:2 Percentile:42.16(Chemistry, Physical)Koshida, Hiroyuki*; Wilde, M.*; Fukutani, Katsuyuki
Journal of Chemical Physics, 160(3), p.034703_1 - 034703_9, 2024/01
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Tei, C.; Otaka, Masahiko; Kuwahara, Daisuke*
Chemical Physics Letters, 829, p.140755_1 - 140755_6, 2023/10
Times Cited Count:1 Percentile:12.81(Chemistry, Physical)We were able to detect the nuclear magnetic resonance (NMR) signal of a liquid sodium clinging to the interface of solid metal particles for the first time. In this study, we confirmed the difference in the relaxation times due to the difference in the interactions between liquid sodium and metal particles suspended in the liquid sodium. It was found that the surface of the micro titanium particles and liquid metallic sodium interact physically, not chemically.
Hirato, Misaki*; Yokoya, Akinari*; Baba, Yuji*; Mori, Seiji*; Fujii, Kentaro*; Wada, Shinichi*; Izumi, Yudai*; Haga, Yoshinori
Physical Chemistry Chemical Physics, 25(21), p.14836 - 14847, 2023/05
Times Cited Count:2 Percentile:28.18(Chemistry, Physical)Nakanishi, Takumi*; Hori, Yuta*; Shigeta, Yasuteru*; Sato, Hiroyasu*; Wu, S.-Q.*; Kiyanagi, Ryoji; Munakata, Koji*; Ohara, Takashi; Sato, Osamu*
Physical Chemistry Chemical Physics, 25(17), p.12394 - 12400, 2023/05
Times Cited Count:3 Percentile:42.17(Chemistry, Physical)Kai, Takeshi; Toigawa, Tomohiro; Ukai, Masatoshi*; Fujii, Kentaro*; Watanabe, Ritsuko*; Yokoya, Akinari*
Journal of Chemical Physics, 158(16), p.164103_1 - 164103_8, 2023/04
Times Cited Count:6 Percentile:63.96(Chemistry, Physical)New insight into water radiolysis and photolysis is indispensable in the dramatic progress of sciences and technologies in various research areas. In the radiation field, reactive hydrated electrons are considerably produced along radiation tracks. Although the formation results from a transient dynamic correlation between ejected electrons and water, the individual mechanisms of electron thermalization, delocalization, and polarization are unknown. Using a dynamic Monte Carlo code, we show herein that the ejected electrons are immediately delocalized by molecular excitations in parallel with phonon polarization and gradually thermalized by momentum transfer with an orientation polarization in a simultaneous manner. Our results show that these mechanisms heavily depend on the intermolecular vibration and rotation modes peculiar to water. We expect our approach to be a powerful technique for connecting physical and chemical processes in various solvents.
 species at SiO/Si interfaces in Si dry oxidation; Comparison between p-Si(001) and n-Si(001) surfaces
species at SiO/Si interfaces in Si dry oxidation; Comparison between p-Si(001) and n-Si(001) surfacesTsuda, Yasutaka; Yoshigoe, Akitaka; Ogawa, Shuichi*; Sakamoto, Tetsuya*; Yamamoto, Yoshiki*; Yamamoto, Yukio*; Takakuwa, Yuji*
Journal of Chemical Physics, 157(23), p.234705_1 - 234705_21, 2022/12
Times Cited Count:2 Percentile:15.57(Chemistry, Physical)Matsuda, Shohei; Nakashima, Nobuaki*; Yokoyama, Keiichi; Taniguchi, Seiji*; Chosrowjan, H.*; Somekawa, Toshihiro*; Yatsuhashi, Tomoyuki*
Chemical Physics Letters, 802, p.139759_1 - 139759_6, 2022/09
Times Cited Count:1 Percentile:6.73(Chemistry, Physical)no abstracts in English
Yamaguchi, Ko*; Kawaguchi, Daisuke*; Miyata, Noboru*; Miyazaki, Tsukasa*; Aoki, Hiroyuki; Yamamoto, Satoru*; Tanaka, Keiji*
Physical Chemistry Chemical Physics, 24(36), p.21578 - 21582, 2022/09
Times Cited Count:13 Percentile:79.00(Chemistry, Physical)Hirade, Tetsuya; Michishio, Koji*; Kobayashi, Yoshinori*; Oshima, Nagayasu*
Chemical Physics Letters, 795, p.139507_1 - 139507_4, 2022/05
Times Cited Count:2 Percentile:6.73(Chemistry, Physical)We obtained the temperature dependence up to 150 C of the triplet positronium (
C of the triplet positronium (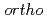 -Ps) lifetime in N,N,N-Trimethyl-N-propylammonium bis(trifluoromethanesulfonyl)imide (TMPA-TFSI) by the vertical slow positron beamline installed at AIST. Positrons penetrate into the liquid surface of TMPA-TFSI with the positron energies of 2 keV and 12 keV to investigate at the near-surface and the balk. The surface structure was visible at 150C, 120C above the melting temperature. The -Ps lifetime became shorter at higher temperatures for both positron energies. Similar temperature dependence had appeared just in water as the result of the reaction of -Ps and radiolysis products such as the OH radicals. The temperature dependence observed for TMPA-TFSI suggested that the chemical reaction of -Ps occurred.
-Ps) lifetime in N,N,N-Trimethyl-N-propylammonium bis(trifluoromethanesulfonyl)imide (TMPA-TFSI) by the vertical slow positron beamline installed at AIST. Positrons penetrate into the liquid surface of TMPA-TFSI with the positron energies of 2 keV and 12 keV to investigate at the near-surface and the balk. The surface structure was visible at 150C, 120C above the melting temperature. The -Ps lifetime became shorter at higher temperatures for both positron energies. Similar temperature dependence had appeared just in water as the result of the reaction of -Ps and radiolysis products such as the OH radicals. The temperature dependence observed for TMPA-TFSI suggested that the chemical reaction of -Ps occurred.
path integral simulationsThomsen, B.; Shiga, Motoyuki
Physical Chemistry Chemical Physics, 24(18), p.10851 - 10859, 2022/05
Times Cited Count:5 Percentile:43.79(Chemistry, Physical)