


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Namie, Masanari; Saito, Junichi
Computational Materials Science, 239, p.112963_1 - 112963_7, 2024/04
Times Cited Count:1 Percentile:30.18(Materials Science, Multidisciplinary)Moriyama, Junichiro*; Takakuwa, Osamu*; Yamaguchi, Masatake; Ogawa, Yuhei*; Tsuzaki, Kaneaki*
Computational Materials Science, 232, p.112650_1 - 112650_11, 2024/01
Times Cited Count:4 Percentile:39.92(Materials Science, Multidisciplinary)The present study focuses on a novel hydrogen-improved strength-ductility balance in some practical Fe-Cr-Ni-based austenitic alloys, which directly depends on the solute hydrogen content. The hydrogen absorption energy of the Fe-Cr-Ni model alloys with the face-centered cubic structure was examined using first-principles calculations to verify the contribution of Cr and Ni substitutions from Fe to the hydrogen solubility in the alloys. The Cr substitution substantially reduced the hydrogen absorption energy compared to the Ni substitution, whereby the increased Cr/Ni ratio exerts higher hydrogen solubility. The propensity in the calculations coincided with the experimental results obtained previously in the practical alloys with various Cr / Ni ratios.
Yamamoto, Yojiro*; Hayakawa, Sho*; Okita, Taira*; Itakura, Mitsuhiro
Computational Materials Science, 229, p.112389_1 - 112389_9, 2023/10
Times Cited Count:2 Percentile:20.33(Materials Science, Multidisciplinary)He bubbles are characteristic microstructures under fusion reactor conditions. They approach and coalesce through their own migration, which significantly impacts the microstructure and material properties. However, these processes, which involve multiple migrations of metal atoms, cannot be treated by molecular dynamics (MD) due to its timescale limitation. In this study, self-evolving atomistic kinetic Monte Carlo (SEAKMC) was used to expand the timescale and reproduce bubble coalescences in Fe. To enhance selections of events that led to the process by avoiding trivial events with an extremely low activation energy such as tiny vibrations of a He atom or short-range displacements of the Fe atom, we introduced two algorithms into SEAKMC, a two-step saddle point search for the former measure and setting a threshold for a displacement distance of the Fe atom for the latter. Furthermore, by adding another algorithm to set an upper bound for the activation energy to prevent selections of events with an impractically high activation energy, we succeeded to reproduce the change in the configuration from dumbbell to elliptical up to a simulated time of 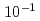 s, 8 orders longer than MD timescales. The developed method is effective for analyzing microstructures of metallic materials containing light elements and is the only method that can reach timescales comparable to those of experiments.
s, 8 orders longer than MD timescales. The developed method is effective for analyzing microstructures of metallic materials containing light elements and is the only method that can reach timescales comparable to those of experiments.
Hayakawa, Sho*; Yamamoto, Yojiro*; Okita, Taira*; Itakura, Mitsuhiro; Suzuki, Katsuyuki*
Computational Materials Science, 218, p.111987_1 - 111987_10, 2023/02
Times Cited Count:1 Percentile:5.58(Materials Science, Multidisciplinary)Tsugawa, Kiyoto*; Hayakawa, Sho*; Okita, Taira*; Aichi, Masaatsu*; Itakura, Mitsuhiro; Suzuki, Katsuyuki*
Computational Materials Science, 215, p.111806_1 - 111806_8, 2022/12
Times Cited Count:6 Percentile:33.27(Materials Science, Multidisciplinary)Tsugawa, Kiyoto*; Hayakawa, Sho*; Iwase, Yuki*; Okita, Taira*; Suzuki, Katsuyuki*; Itakura, Mitsuhiro; Aichi, Masaatsu*
Computational Materials Science, 210, p.111450_1 - 111450_9, 2022/07
Times Cited Count:18 Percentile:75.48(Materials Science, Multidisciplinary)Tsuru, Tomohito; Itakura, Mitsuhiro; Yamaguchi, Masatake; Watanabe, Chihiro*; Miura, Hiromi*
Computational Materials Science, 203, p.111081_1 - 111081_9, 2022/02
Times Cited Count:17 Percentile:64.50(Materials Science, Multidisciplinary)The deformation mode of some titanium (Ti) alloys differs from that of pure Ti due to the presence of alloying elements in  -phase. Herein, we investigated all possible slip modes in pure Ti and the effects of Al and V solutes as typical additive elements on the dislocation motion in -Ti alloys using density functional theory (DFT) calculations. The stacking fault (SF) energies in possible slip planes indicated that both Al and V solutes reduce the SF energy in the basal plane and, in contrast, the Al solute increases the SF energy particularly in the prismatic plane. DFT calculations were subsequently performed to simulate dislocation core structures. The energy landscape of the transition between all possible dislocation core structures and the barriers for dislocation glide in various slip planes clarified the nature of dislocation motion in pure Ti. (i) the energy of prismatic core is higher than most stable pyramidal core, and thereby dislocations need to overcome the energy barrier of the cross-slip (22.8 meV/b) when they move in the prismatic plane, (ii) the energy difference between the prismatic and basal cores is larger (127 meV/b), that indicates the basal slip does not activate, (iii) however, the Peierls barrier for motion in the basal plane is not as high (16 meV/b). Direct calculations for the dislocation core around solutes revealed that both Al and V solutes facilitate dislocation motion in the basal plane by reducing the energy difference between the prismatic and basal cores. The effect of solutes characterizes the difference in the deformation mode of pure Ti and -Ti alloys.
-phase. Herein, we investigated all possible slip modes in pure Ti and the effects of Al and V solutes as typical additive elements on the dislocation motion in -Ti alloys using density functional theory (DFT) calculations. The stacking fault (SF) energies in possible slip planes indicated that both Al and V solutes reduce the SF energy in the basal plane and, in contrast, the Al solute increases the SF energy particularly in the prismatic plane. DFT calculations were subsequently performed to simulate dislocation core structures. The energy landscape of the transition between all possible dislocation core structures and the barriers for dislocation glide in various slip planes clarified the nature of dislocation motion in pure Ti. (i) the energy of prismatic core is higher than most stable pyramidal core, and thereby dislocations need to overcome the energy barrier of the cross-slip (22.8 meV/b) when they move in the prismatic plane, (ii) the energy difference between the prismatic and basal cores is larger (127 meV/b), that indicates the basal slip does not activate, (iii) however, the Peierls barrier for motion in the basal plane is not as high (16 meV/b). Direct calculations for the dislocation core around solutes revealed that both Al and V solutes facilitate dislocation motion in the basal plane by reducing the energy difference between the prismatic and basal cores. The effect of solutes characterizes the difference in the deformation mode of pure Ti and -Ti alloys.
Okita, Taira*; Terayama, Satoshi*; Tsugawa, Kiyoto*; Kobayashi, Keita; Okumura, Masahiko; Itakura, Mitsuhiro; Suzuki, Katsuyuki*
Computational Materials Science, 202, p.110865_1 - 110865_9, 2022/02
Times Cited Count:7 Percentile:33.32(Materials Science, Multidisciplinary)Terayama, Satoshi*; Iwase, Yuki*; Hayakawa, Sho*; Okita, Taira*; Itakura, Mitsuhiro; Suzuki, Katsuyuki*
Computational Materials Science, 195, p.110479_1 - 110479_12, 2021/07
Times Cited Count:13 Percentile:48.84(Materials Science, Multidisciplinary)Kobayashi, Keita; Nakamura, Hiroki; Yamaguchi, Akiko; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Computational Materials Science, 188, p.110173_1 - 110173_14, 2021/02
Times Cited Count:25 Percentile:75.68(Materials Science, Multidisciplinary)no abstracts in English
Yamaguchi, Masatake; Ebihara, Kenichi; Itakura, Mitsuhiro; Tsuru, Tomohito; Matsuda, Kenji*; Toda, Hiroyuki*
Computational Materials Science, 156, p.368 - 375, 2019/01
Times Cited Count:50 Percentile:84.06(Materials Science, Multidisciplinary)The segregation of multiple hydrogen atoms along aluminum (Al) grain boundaries (GBs) and fracture surfaces (FSs) was investigated through first-principles calculations considering the characteristics of GBs. The results indicate that hydrogen segregation is difficult along low-energy GBs. The segregation energy of multiple hydrogen atoms along GBs and FSs and the cohesive energy was obtained for three types of high-energy Al GBs. With increasing hydrogen segregation along the GBs, the cohesive energy of the GB decreases and approaches zero with no decrease in GB segregation energy. The GB cohesive energy decreases in parallel with the volume expansion of the region of low electron density along the GB.
 precipitate in Al-Mg-Zn alloys
precipitate in Al-Mg-Zn alloysTsuru, Tomohito; Yamaguchi, Masatake; Ebihara, Kenichi; Itakura, Mitsuhiro; Shiihara, Yoshinori*; Matsuda, Kenji*; Toda, Hiroyuki*
Computational Materials Science, 148, p.301 - 306, 2018/06
Times Cited Count:57 Percentile:84.59(Materials Science, Multidisciplinary)Hydrogen embrittlement susceptibility of high strength 7xxx series Al alloys has been recognized as the critical issues in the practical use of Al alloys. Focusing on the interface between MgZn precipitates and an Al matrix, which is considered as one of the important segregation sites in these alloys, we investigated the stable 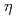 -MgZn-Al interface, and the possible hydrogen trap sites in MgZn and at the -MgZn-Al interface via first-principles calculation. Most of the interstitial sites inside the MgZn crystal were not possible trap sites because their energy is relatively higher than that of other trap sites. The trap energy of the most favorable site at the -MgZn-Al is approximately -0.3 eV/H, which is more stable that of the interstitial site at the grain boundary. The interface between MgZn and Al is likely to be a possible trap site in Al alloys.
-MgZn-Al interface, and the possible hydrogen trap sites in MgZn and at the -MgZn-Al interface via first-principles calculation. Most of the interstitial sites inside the MgZn crystal were not possible trap sites because their energy is relatively higher than that of other trap sites. The trap energy of the most favorable site at the -MgZn-Al is approximately -0.3 eV/H, which is more stable that of the interstitial site at the grain boundary. The interface between MgZn and Al is likely to be a possible trap site in Al alloys.
O and the reaction of Fe + ONakazawa, Tetsuya
Computational Materials Science, 146, p.334 - 345, 2018/04
Times Cited Count:2 Percentile:6.41(Materials Science, Multidisciplinary)Density functional theory calculations are performed on all possible structures of FeO using the hybrid B3LYP functional and the B3LYP functional combined with the broken-symmetry (BS) approach to obtain the most stable isomers. Based on the obtained stable isomers, the reaction mechanism of Fe + O toward rhombic Fe(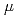 -O) is considered. The BS-singlet state of the rhombic Fe(-O) 2.1 is found to be the ground state of all FeO isomers. The
-O) is considered. The BS-singlet state of the rhombic Fe(-O) 2.1 is found to be the ground state of all FeO isomers. The  A" state of the open-cycle (
A" state of the open-cycle (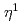 -O) Fe(-O) 2.11 and
-O) Fe(-O) 2.11 and  A state of the near-linear OFeOFe 1.4 are found to have the second and third lowest energy states. The lowest-lying energy states of the bare FeO clusters do not favor three-dimensional structures, but favor the linear and planar structures. The singlet and nonet energy surfaces calculated for the reaction of Fe + O toward rhombic Fe(-O) suggest that the reaction adiabatically proceeds with spin inversion from the nonet to singlet state.
A state of the near-linear OFeOFe 1.4 are found to have the second and third lowest energy states. The lowest-lying energy states of the bare FeO clusters do not favor three-dimensional structures, but favor the linear and planar structures. The singlet and nonet energy surfaces calculated for the reaction of Fe + O toward rhombic Fe(-O) suggest that the reaction adiabatically proceeds with spin inversion from the nonet to singlet state.
with ONakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 117, p.455 - 467, 2016/05
Times Cited Count:8 Percentile:26.08(Materials Science, Multidisciplinary)The reactions of Fe and FeO with O and the products of these reactions are investigated at the B3LYP/6-311+G(d) level. The reactions are considered in terms of the calculated potential energy surfaces, the interaction energies between reactant species, and the energies required to populate the higher electronic states such as the excited states of Fe(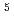 F, F) and O(
F, F) and O(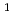
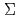
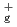 ). It is found that the reactions of Fe with O are endothermic and that the direct formation of dioxide OFeO is due to an ionic interaction of Fe
). It is found that the reactions of Fe with O are endothermic and that the direct formation of dioxide OFeO is due to an ionic interaction of Fe with O
with O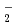 . Furthermore, the diabatic transitions from the covalent and ionic surfaces onto another ionic surface with energy barriers allow the Fe + O reaction to proceed toward the formation of OFeO. There are other paths corresponding to the vertical excitation from peroxide Fe(O) to dioxide OFeO. The OFeO + O reactions are found to endothermically produce the 2- and -(O)FeO complexes, and the low-lying states of complexes are found to be closely located in energy. The CCSD and CCSD(T) single point energy calculations are performed with the structures optimized at the B3LYP level. The results of NBO and Mulliken analyses and the harmonic vibrational frequencies are presented.
. Furthermore, the diabatic transitions from the covalent and ionic surfaces onto another ionic surface with energy barriers allow the Fe + O reaction to proceed toward the formation of OFeO. There are other paths corresponding to the vertical excitation from peroxide Fe(O) to dioxide OFeO. The OFeO + O reactions are found to endothermically produce the 2- and -(O)FeO complexes, and the low-lying states of complexes are found to be closely located in energy. The CCSD and CCSD(T) single point energy calculations are performed with the structures optimized at the B3LYP level. The results of NBO and Mulliken analyses and the harmonic vibrational frequencies are presented.
-ironAbe, Yosuke
Computational Materials Science, 74, p.23 - 26, 2013/06
Times Cited Count:9 Percentile:29.01(Materials Science, Multidisciplinary)The diffusion process of a single self-interstitial atom (SIA) in -iron was studied by a hyper-molecular dynamics (hyper-MD) method that has been demonstrated to efficiently accelerate slow diffusive conformational transitions that occur in various materials. By adding a local bias potential, the acceleration of the dynamics becomes significantly higher at lower temperatures, e.g., on the order of 5 at 300 K, without changing the dynamics of the diffusion process as calculated using a conventional MD simulation. The validity of the results demonstrates that the hyper-MD method with an appropriate local bias potential can be applicable for determination of the diffusion process of SIAs in bulk materials.
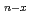 Si
Si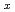 (
(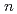 ,
, 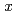
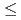 6) clusters; Theoretical investigation based on density functional theory
6) clusters; Theoretical investigation based on density functional theoryNakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 68, p.350 - 360, 2013/02
Times Cited Count:4 Percentile:14.17(Materials Science, Multidisciplinary)Based on DFT calculations, FeSi (, 6) clusters are investigated. The Si substitutions increase cohesion of Fe cluster. The substitutions affect structural parameter, bond strength, and magnetic moment. The changes are connected with electron transfer from Fe to Si atoms.
Mo (, 6) clustersNakazawa, Tetsuya; Kaji, Yoshiyuki
Computational Materials Science, 55, p.365 - 375, 2012/04
Times Cited Count:12 Percentile:35.44(Materials Science, Multidisciplinary) tilt grain boundary using multi-phase-field model with penalty for multiple junctions
tilt grain boundary using multi-phase-field model with penalty for multiple junctionsHirouchi, Tomoyuki*; Tsuru, Tomohito; Shibutani, Yoji*
Computational Materials Science, 53(1), p.474 - 482, 2012/02
Times Cited Count:23 Percentile:54.75(Materials Science, Multidisciplinary)Multi-phase-field (MPF) model with a higher-order term representing energetic penalty for multiple junctions was proposed to predict the grain growth accompanying the inclination dependence of grain boundary (GB) energy and mobility. The inclination effect was introduced on the basis of GB energy obtained from molecular dynamics simulations. The preliminary grain growth simulation of an isolated grain surrounded by 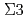 GB certified that the analytical equilibrium shape was well reproduced. The augmented higher-order term added to conventional MPF model could improve convergence and stability of numerical calculations around triple junction (TJ) region even if there exists the large GB energy gap at the TJ. For the polycrystalline grain growth simulations with the GB energy distribution according to the misorientation angle of Al tilt GB, GB inclination lead the weak anisotropy characterized by
GB certified that the analytical equilibrium shape was well reproduced. The augmented higher-order term added to conventional MPF model could improve convergence and stability of numerical calculations around triple junction (TJ) region even if there exists the large GB energy gap at the TJ. For the polycrystalline grain growth simulations with the GB energy distribution according to the misorientation angle of Al tilt GB, GB inclination lead the weak anisotropy characterized by 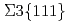 twin boundary.
twin boundary.
Igarashi, Takahiro; Nakazawa, Tetsuya; Suzuki, Chikashi; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 50(12), p.3346 - 3349, 2011/12
Times Cited Count:1 Percentile:4.27(Materials Science, Multidisciplinary)The authors have developed a new approach for large-scale systems including over 100,000 atoms to obtain physical strength from the viewpoint of atom-atom bonding energy. In this study, the authors applied this method to SiH molecule and crystalline silica system, and carried out bond order and bonding energy analyses. In this analysis, the developed method offered almost the same analytical accuracy as the first principle method, while its calculation speed was much faster than that of the latter.
molecule and crystalline silica system, and carried out bond order and bonding energy analyses. In this analysis, the developed method offered almost the same analytical accuracy as the first principle method, while its calculation speed was much faster than that of the latter.
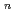 clusters (n 6) with Cr substitutions; UB3LYP/LanL2DZ calculation
clusters (n 6) with Cr substitutions; UB3LYP/LanL2DZ calculationNakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 50(3), p.982 - 990, 2011/01
Times Cited Count:10 Percentile:31.89(Materials Science, Multidisciplinary)Geometric parameters, binding energies, natural populations, natural electron configurations and magnetic moments are obtained for the clusters of Fe, Cr and FeCr ( = 2-6, = 1-6) optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The substitutional effects of Cr in Fen are found in the properties. The binding energies of Fen are generally decreased by successive substitutions of Cr for Fe atoms in the clusters. In the mixed Fe-Cr clusters most of Cr-Cr bond lengths are larger than the Fe-Fe and Fe-Cr bond lengths. Among the Fe-Fe, Fe-Cr and Cr-Cr bond lengths in the mixed clusters, the trend is found to become larger in that order. The larger distances between atoms in the mixed clusters are mostly caused by the strong repulsion due to magnetic frustration between atoms. The changes are associated with those of electronic structure by the Cr substitutions, especially with the extent of contribution of 4s and 3d electrons to bond.
Nakazawa, Tetsuya; Igarashi, Takahiro; Tsuru, Tomohito; Kaji, Yoshiyuki
Computational Materials Science, 46(2), p.367 - 375, 2009/08
Times Cited Count:26 Percentile:59.21(Materials Science, Multidisciplinary)The clusters of Fe, Ni, and Fe-Ni are investigated computationally using a density functional approach. The geometries of clusters are optimized under the constraint of well-defined point group symmetries at the UB3LYP/LanL2DZ level. The equilibrium geometries and binding energies are presented and discussed, together with natural populations and natural electron configurations. In addition, the binding energies of FeNi clusters are found to generally decrease by successive substitutions of Ni atoms for Fe atoms. For Fe Ni clusters, the comparisons on total energies between isomers indicate that Ni atoms energetically prefer clustering in the mixed Fe-Ni clusters. The calculations for FeNi clusters show that the clustering leads to a segregation of Ni atoms from Fe atoms.
