


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
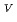 O
O
 /U
/U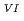 O
O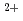 redox equilibrium in [emim]TfN-based liquid electrolytes towards uranium redox-flow battery
redox equilibrium in [emim]TfN-based liquid electrolytes towards uranium redox-flow batteryTakao, Koichiro*; Ouchi, Kazuki; Komatsu, Atsushi; Kitatsuji, Yoshihiro; Watanabe, Masayuki
European Journal of Inorganic Chemistry, 27(14), p.e202300787_1 - e202300787_7, 2024/05
Times Cited Count:1 Percentile:0.00(Chemistry, Inorganic & Nuclear)Electrochemical behavior of U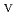 O/U
O/U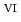 O in 1-ethyl-3-methylimidazolium bis(trifluoromethyl)sulfonylamide ([emim]TfN) ionic liquid was studied to clarify what are required to attain its redox reversibility for utilizing depleted U as an electrode active material in a redox flow battery. As a result, reversibility of the UO/UO redox reaction was successfully achieved in use of a glassy carbon working electrode under presence of Cl
O in 1-ethyl-3-methylimidazolium bis(trifluoromethyl)sulfonylamide ([emim]TfN) ionic liquid was studied to clarify what are required to attain its redox reversibility for utilizing depleted U as an electrode active material in a redox flow battery. As a result, reversibility of the UO/UO redox reaction was successfully achieved in use of a glassy carbon working electrode under presence of Cl in [emim]TfN. To improve diffusivity of solutes, [emim]TfN diluted with an auxiliary molecular solvent, N,N-dimethylformamide (DMF). We have succeeded in demonstrating a reversible redox reaction of [UOCl
in [emim]TfN. To improve diffusivity of solutes, [emim]TfN diluted with an auxiliary molecular solvent, N,N-dimethylformamide (DMF). We have succeeded in demonstrating a reversible redox reaction of [UOCl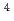 ]
]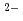 + e = [UOCl]
+ e = [UOCl]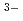 in the 50:50 v/v [emim]TfN-DMF liquid electrolyte containing Cl.
in the 50:50 v/v [emim]TfN-DMF liquid electrolyte containing Cl.
O -75MoO binary glasses composed of isolated MoO
-75MoO binary glasses composed of isolated MoO
Masuno, Atsunobu*; Munakata, Sae*; Okamoto, Yoshihiro; Yaji, Toyonari*; Kosugi, Yoshihisa*; Shimakawa, Yuichi*
Inorganic Chemistry, 63(12), p.5701 - 5708, 2024/03
Times Cited Count:2 Percentile:65.79(Chemistry, Inorganic & Nuclear)Transparent and brown LaO-MoO binary glasses were prepared in bulk form using a levitation technique. The glass-forming range was limited, with the primary composition being approximately 25 mol% LaO. This glass exhibited a clear crystallization at 546  C, while determining its glass transition temperature was difficult. Notably, despite its amorphous nature, the glass possessed a density and packing density comparable to those of crystalline LaMoO
C, while determining its glass transition temperature was difficult. Notably, despite its amorphous nature, the glass possessed a density and packing density comparable to those of crystalline LaMoO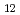 . X-ray absorption fine structure and Raman scattering analyses revealed that the glass structure closely resembles LaMoO due to the presence of isolated MoO units, whereas disordered atomic arrangement around La atoms was confirmed. The glass demonstrated transparency ranging from 378 to 5500 nm, and the refractive index at 1.0
. X-ray absorption fine structure and Raman scattering analyses revealed that the glass structure closely resembles LaMoO due to the presence of isolated MoO units, whereas disordered atomic arrangement around La atoms was confirmed. The glass demonstrated transparency ranging from 378 to 5500 nm, and the refractive index at 1.0 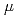 m was estimated to be 2.0. The optical bandgap energy was 3.46 eV, which was slightly smaller than that of LaMoO. Additionally, the glass displayed a transparent region ranging from 6.5 to 8.0 m. This occurrence results from the decreased diversity of MoO
m was estimated to be 2.0. The optical bandgap energy was 3.46 eV, which was slightly smaller than that of LaMoO. Additionally, the glass displayed a transparent region ranging from 6.5 to 8.0 m. This occurrence results from the decreased diversity of MoO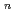 units and connectivity of Mo-O-Mo, which resulted in the reduced overlap of multiphonon absorption. This glass formation, with its departure from conventional glass-forming rules, resulted in distinctive glasses with crystal-like atomic arrangements.
units and connectivity of Mo-O-Mo, which resulted in the reduced overlap of multiphonon absorption. This glass formation, with its departure from conventional glass-forming rules, resulted in distinctive glasses with crystal-like atomic arrangements.
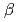 -MoO whiskers in
-MoO whiskers in 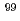 Mo/
Mo/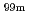 Tc radioisotope production and Mo/Tc extraction using hot atoms
Tc radioisotope production and Mo/Tc extraction using hot atomsNgo, M. C.*; Fujita, Yoshitaka; Suzuki, Tatsuya*; Do, T. M. D.*; Seki, Misaki; Nakayama, Tadachika*; Niihara, Koichi*; Suematsu, Hisayuki*
Inorganic Chemistry, 62(32), p.13140 - 13147, 2023/08
Times Cited Count:5 Percentile:54.36(Chemistry, Inorganic & Nuclear)Technetium-99m (Tc) is one of the most important radioisotopes for diagnostic radio-imaging applications. Tc is a daughter product of the Mo isotope. There are two methods used to produce Mo/Tc: the nuclear fission (n,f) and the neutron capture (n,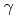 ) methods. Between them, the (n,f) method is the main route, used for approximately 90% of the world's production. However, the (n,f) method faces numerous problems, including the use of highly enriched uranium, the release of highly radioactive waste, and nonproliferation problems. Therefore, the (n,) method is being developed as a future replacement for the (n,f) method. In this work, -MoO whiskers prepared by the thermal evaporation method and
) methods. Between them, the (n,f) method is the main route, used for approximately 90% of the world's production. However, the (n,f) method faces numerous problems, including the use of highly enriched uranium, the release of highly radioactive waste, and nonproliferation problems. Therefore, the (n,) method is being developed as a future replacement for the (n,f) method. In this work, -MoO whiskers prepared by the thermal evaporation method and  -MoO particles were irradiated in a nuclear reactor to produce Mo/Tc via neutron capture. The irradiated targets were dispersed into water to extract the Mo/Tc. As a result, -MoO whisker yielded higher Mo extraction rate than that from -MoO. In addition, by comparing the dissolved
-MoO particles were irradiated in a nuclear reactor to produce Mo/Tc via neutron capture. The irradiated targets were dispersed into water to extract the Mo/Tc. As a result, -MoO whisker yielded higher Mo extraction rate than that from -MoO. In addition, by comparing the dissolved 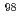 Mo concentrations in water, we clarified a prominent hot-atom of -MoO whiskers. This research is the first demonstration of -MoO being used as an irradiation target in the neutron capture method. On the basis of the results, -MoO is considered a promising irradiation target for producing Mo/Tc by neutron capture and using water for the radioisotope extraction process in the future.
Mo concentrations in water, we clarified a prominent hot-atom of -MoO whiskers. This research is the first demonstration of -MoO being used as an irradiation target in the neutron capture method. On the basis of the results, -MoO is considered a promising irradiation target for producing Mo/Tc by neutron capture and using water for the radioisotope extraction process in the future.
Hirata, Sakiko*; Kusaka, Ryoji; Meiji, Shogo*; Tamekuni, Seita*; Okudera, Kosuke*; Hamada, Shoken*; Sakamoto, Chihiro*; Honda, Takumi*; Matsushita, Kosuke*; Muramatsu, Satoru*; et al.
Inorganic Chemistry, 62(1), p.474 - 486, 2023/01
Times Cited Count:3 Percentile:28.83(Chemistry, Inorganic & Nuclear)Zheng, X.*; Kato, Masaru*; Uemura, Yohei*; Matsumura, Daiju; Yagi, Ichizo*; Takahashi, Kiyonori*; Noro, Shinichiro*; Nakamura, Takayoshi*
Inorganic Chemistry, 62(3), p.1257 - 1263, 2023/01
Times Cited Count:8 Percentile:79.46(Chemistry, Inorganic & Nuclear)Yamagata, Kazuhito*; Ouchi, Kazuki; Marumo, Kazuki*; Tasaki-Handa, Yuiko*; Haraga, Tomoko; Saito, Shingo*
Inorganic Chemistry, 62(2), p.730 - 738, 2023/01
Times Cited Count:3 Percentile:42.77(Chemistry, Inorganic & Nuclear)The inert NpO complex with a fluorescein-modified phenanthroline-2,9-dicarboxylic acid was found by kinetic selection using polyacrylamide gel electrophoresis (PAGE) from a small chemical library. The small spontaneous dissociation rate constant of 8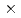 10
10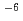 s
s (the half-life of 23 hours) was determined. This is the singly-charged NpO complex exhibiting unusual kinetic inertness in aqueous solution, one million times slower than widely accepted fast kinetics of neptunyl complexes. Selective fluorescence detection of NpO was achieved in PAGE with a detection limit of 68 pmol dm
(the half-life of 23 hours) was determined. This is the singly-charged NpO complex exhibiting unusual kinetic inertness in aqueous solution, one million times slower than widely accepted fast kinetics of neptunyl complexes. Selective fluorescence detection of NpO was achieved in PAGE with a detection limit of 68 pmol dm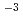 (17 fg). This system was successfully applied to simulated spent nuclear fuel and high-level radioactive waste samples.
(17 fg). This system was successfully applied to simulated spent nuclear fuel and high-level radioactive waste samples.
-edge to assess the U(V) electronic structure in FeUOYomogida, Takumi; Akiyama, Daisuke*; Ouchi, Kazuki; Kumagai, Yuta; Higashi, Kotaro*; Kitatsuji, Yoshihiro; Kirishima, Akira*; Kawamura, Naomi*; Takahashi, Yoshio*
Inorganic Chemistry, 61(50), p.20206 - 20210, 2022/12
Times Cited Count:7 Percentile:59.79(Chemistry, Inorganic & Nuclear)FeUO was studied to clarify the electronic structure of U(V) in a metal monouranate compound. We obtained the peak splitting of HERFD-XANES spectra utilizing high-energy-resolution fluorescence detection-X-ray absorption near edge structure (HERFD-XANES) spectroscopy at the U L-edge, which is a novel technique in the U(V) compounds. Theoretical calculations revealed that the peak splitting was caused by splitting the 6d orbital of U(V). Such distinctive electronic states are of major interest to researchers and engineers working in various fields, from fundamental physics to the nuclear industry and environmental sciences for actinide elements.
O-pentadentate planar ligands designed for the strongest and selective capture of uranium from seawaterMizumachi, Takumi*; Sato, Minami*; Kaneko, Masashi; Takeyama, Tomoyuki*; Tsushima, Satoru*; Takao, Koichiro*
Inorganic Chemistry, 61(16), p.6175 - 6181, 2022/04
Times Cited Count:6 Percentile:53.89(Chemistry, Inorganic & Nuclear)Based on unique 5-fold equatorial coordination of UO, water-compatible pentadentate planar ligands, Hsaldian and its derivatives, were designed as strong and selective capture of UO in seawater. In the simulated seawater condition (0.5 M NaCl + 2.3 mM HCO/CO, pH 8), saldian shows the strongest complexation with UO to form UO(saldian) (log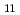 = 28.05
= 28.05 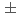 0.07), which is more than 10 order of magnitude greater than amidoxime-based or -inspired ligand systems most commonly employed for U capture from seawater. Good selectivity for UO from other metal ions coexisting in seawater was also demonstrated.
0.07), which is more than 10 order of magnitude greater than amidoxime-based or -inspired ligand systems most commonly employed for U capture from seawater. Good selectivity for UO from other metal ions coexisting in seawater was also demonstrated.
TaO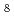
Kimura, Kenta*; Yagi, Naoki*; Hasegawa, Shunsuke*; Hagihara, Masato; Miyake, Atsushi*; Tokunaga, Masashi*; Cao, H.*; Masuda, Takatsugu*; Kimura, Tsuyoshi*
Inorganic Chemistry, 60(20), p.15078 - 15084, 2021/10
Times Cited Count:3 Percentile:22.10(Chemistry, Inorganic & Nuclear)Simonnet, M.; Kobayashi, Toru; Shimojo, Kojiro; Yokoyama, Keiichi; Yaita, Tsuyoshi
Inorganic Chemistry, 60(17), p.13409 - 13418, 2021/09
Times Cited Count:25 Percentile:91.54(Chemistry, Inorganic & Nuclear)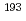 Ir M
Ir M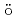 ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adducts
ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adductsKaneko, Masashi; Nakashima, Satoru*
Inorganic Chemistry, 60(17), p.12740 - 12752, 2021/09
Times Cited Count:4 Percentile:29.88(Chemistry, Inorganic & Nuclear)In the present study, density functional theory (DFT) calculation was applied to Vaska's complexes of formula 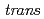 -[IrCl(CO)(PPh)], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
-[IrCl(CO)(PPh)], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
Kwon, H.*; Pietrasiak, E.*; Ohara, Takashi; Nakao, Akiko*; Chae, B.*; Hwang, C.-C.*; Jung, D.*; Hwang, I.-C.*; Ko, Y. H.*; Kim, K.*; et al.
Inorganic Chemistry, 60(9), p.6403 - 6409, 2021/05
Times Cited Count:0 Percentile:0.00(Chemistry, Inorganic & Nuclear)Nakano, Satoshi*; Sano, Asami; Hattori, Takanori; Machida, Shinichi*; Komatsu, Kazuki*; Fujihisa, Hiroshi*; Yamawaki, Hiroshi*; Goto, Yoshito*; Kikegawa, Takumi*
Inorganic Chemistry, 60(5), p.3065 - 3073, 2021/03
Times Cited Count:13 Percentile:73.97(Chemistry, Inorganic & Nuclear)X-ray and neutron diffraction analyses of ammonia borane were conducted at ambient and high pressures. The H-H distance in dihydrogen bonds was shorter than twice the van der Waals radius (2.4 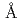 ). The half of the dihydrogen bonds were broken on phase transition from AP to the first high pressure phase (HP1) at approximately 1.2 GPa as revealed by an increase in the H-H distances. On further pressure increase, all of the H-H distances became shorter than 2.4 again, implying the pressure-induced reformation of the dihydrogen bonds. Furthermore, the HP1 transformed to the second one with the structure of
). The half of the dihydrogen bonds were broken on phase transition from AP to the first high pressure phase (HP1) at approximately 1.2 GPa as revealed by an increase in the H-H distances. On further pressure increase, all of the H-H distances became shorter than 2.4 again, implying the pressure-induced reformation of the dihydrogen bonds. Furthermore, the HP1 transformed to the second one with the structure of 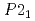 (Z = 2) at about 11 GPa. In this phase transition, the inclination of the molecule axis became larger and the number of types of dihydrogen bonds increased from 6 to 11. Just before the third transition at 18.9 GPa, the shortest dihydrogen bond decreased to 1.65 . The present study experimentally first confirmed the breakage and reformation of the dihydrogen bonds by the structural change under pressure.
(Z = 2) at about 11 GPa. In this phase transition, the inclination of the molecule axis became larger and the number of types of dihydrogen bonds increased from 6 to 11. Just before the third transition at 18.9 GPa, the shortest dihydrogen bond decreased to 1.65 . The present study experimentally first confirmed the breakage and reformation of the dihydrogen bonds by the structural change under pressure.
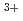 complexes of mixed N,O-donor ligands demonstrated on a 10-fold [Eu(TPAMEN)] chelate complex
complexes of mixed N,O-donor ligands demonstrated on a 10-fold [Eu(TPAMEN)] chelate complexSchnaars, K.; Kaneko, Masashi; Fujisawa, Kiyoshi*
Inorganic Chemistry, 60(4), p.2477 - 2491, 2021/02
Times Cited Count:6 Percentile:44.28(Chemistry, Inorganic & Nuclear)To reduce high-level radiotoxic waste generated by nuclear power plants, highly selective separation agents for minor actinides are mandatory. The mixed N,O-donor ligand 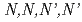 -tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (HTPAEN) has shown good performance as a masking agent in Am/Eu separation studies. In this work, we examine whether a decrease in O-donor basicity can promote the M-N
-tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (HTPAEN) has shown good performance as a masking agent in Am/Eu separation studies. In this work, we examine whether a decrease in O-donor basicity can promote the M-N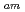 interactions. Therefore, we replace the deprotonated "charged" carboxylic acid groups of TPAEN
interactions. Therefore, we replace the deprotonated "charged" carboxylic acid groups of TPAEN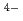 by neutral amide groups and introduce -tetrakis[(6-
by neutral amide groups and introduce -tetrakis[(6-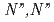 -diethylcarbamoylpyridin-2-yl)methyl]ethylenediamine (TPAMEN) as a new ligand. TPAMEN was crystallized with Eu(OTf) and Eu(NO) 6HO to form positively charged 1:1 [Eu(TPAMEN)] complexes in the solid state. Alterations in the M-O/N bond distances are compared to [Eu(TPAEN)] and investigated by DFT calculations to expose the differences in charge/energy density distributions at europium(III) and the donor functionalities of the TPAEN and TPAMEN. On the basis of estimations of the bond orders, atomic charges spin populations, and density of states in the Eu and potential Am and Cm complexes, the specific contributions of the donor-metal interaction are analyzed. The prediction of complex formation energy differences for the [M(TPAEN)] and [M(TPAMEN)] (M = Eu, Am) complexes provide an outlook on the potential performance of TPAMEN in Am/Eu separation.
-diethylcarbamoylpyridin-2-yl)methyl]ethylenediamine (TPAMEN) as a new ligand. TPAMEN was crystallized with Eu(OTf) and Eu(NO) 6HO to form positively charged 1:1 [Eu(TPAMEN)] complexes in the solid state. Alterations in the M-O/N bond distances are compared to [Eu(TPAEN)] and investigated by DFT calculations to expose the differences in charge/energy density distributions at europium(III) and the donor functionalities of the TPAEN and TPAMEN. On the basis of estimations of the bond orders, atomic charges spin populations, and density of states in the Eu and potential Am and Cm complexes, the specific contributions of the donor-metal interaction are analyzed. The prediction of complex formation energy differences for the [M(TPAEN)] and [M(TPAMEN)] (M = Eu, Am) complexes provide an outlook on the potential performance of TPAMEN in Am/Eu separation.
Narita, Hirokazu*; Kasuya, Ryo*; Suzuki, Tomoya*; Motokawa, Ryuhei; Tanaka, Mikiya*
Encyclopedia of Inorganic and Bioinorganic Chemistry (Internet), 28 Pages, 2020/12
Tamain, C.*; Bonato, L.*; Aupiais, J.*; Dumas, T.*; Guillaumont, D.*; Barkleit, A.*; Berthon, C.*; Solari, P. L.*; Ikeda, Atsushi; Guilbard, P.*; et al.
European Journal of Inorganic Chemistry, 2020(14), p.1331 - 1344, 2020/04
Times Cited Count:4 Percentile:24.09(Chemistry, Inorganic & Nuclear)The molecular characterization based on multi-technique approach has led to major highlights on revealing the coordination environment of americium (Am) surrounded by citric acid (HCitH). The structure of the different complexes at pH 1 and 3 are described. These characterizations are made possible by the comparison of the americium-citric acid system with the americium-tricarballylic acid (one analogue of the citric acid without the alpha-hydroxo group).
Ru Mssbauer parameters of ruthenium-nitrosyl complexesKaneko, Masashi; Kato, Akane*; Nakashima, Satoru*; Kitatsuji, Yoshihiro
Inorganic Chemistry, 58(20), p.14024 - 14033, 2019/10
Times Cited Count:12 Percentile:56.50(Chemistry, Inorganic & Nuclear)We applied density functional theory calculations to ruthenium-nitrosyl complexes, which are known to exist in high-level radioactive waste, to give a theoretical correlation between Ru Mssbauer spectroscopic parameters (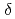 and
and 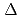
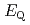 ) and ligand field strength (
) and ligand field strength (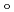 ) for the first time. The structures of the series of complexes, [Ru(NO)L
) for the first time. The structures of the series of complexes, [Ru(NO)L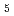 ] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO)L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C
] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO)L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C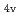 symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of
symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of 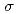 -donor and
-donor and 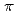 -acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
-acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
Narita, Hirokazu*; Nicolson, R. M.*; Motokawa, Ryuhei; Ito, Fumiyuki*; Morisaku, Kazuko*; Goto, Midori*; Tanaka, Mikiya*; Heller, W. T.*; Shiwaku, Hideaki; Yaita, Tsuyoshi; et al.
Inorganic Chemistry, 58(13), p.8720 - 8734, 2019/07
Times Cited Count:20 Percentile:75.87(Chemistry, Inorganic & Nuclear)Yamaguchi, Toshio*; Nishino, Masaaki*; Yoshida, Koji*; Takumi, Masaharu*; Nagata, Kiyofumi*; Hattori, Takanori
European Journal of Inorganic Chemistry, 2019(8), p.1170 - 1177, 2019/02
Times Cited Count:18 Percentile:67.90(Chemistry, Inorganic & Nuclear)Neutron diffraction measurements of an aqueous 2 mol dm CaCl solutions in DO have been made at 1 GPa, 298 K as well as 0.1 MPa, 298 K. The experimental structure factors are subjected to Empirical Potential Structure Refinement (EPSR) modeling to reveal the ion hydration and association and solvent water at the atomic level. About seven water molecules surround Ca at the Ca-O and Ca-D distances of 2.44 and 3.70 , respectively, at both pressures, suggesting no significant pressure effect on the cation hydration. On the other hand, the Cl ion shows a drastic change in water oxygen coordination from 7 at 0.1 MPa to 14 at 1 GPa, accompanied by shortening of Cl-O distance from 3.18 to 3.15 . However, the number of water hydrogen atoms around Cl does not change significantly as 6.0
solutions in DO have been made at 1 GPa, 298 K as well as 0.1 MPa, 298 K. The experimental structure factors are subjected to Empirical Potential Structure Refinement (EPSR) modeling to reveal the ion hydration and association and solvent water at the atomic level. About seven water molecules surround Ca at the Ca-O and Ca-D distances of 2.44 and 3.70 , respectively, at both pressures, suggesting no significant pressure effect on the cation hydration. On the other hand, the Cl ion shows a drastic change in water oxygen coordination from 7 at 0.1 MPa to 14 at 1 GPa, accompanied by shortening of Cl-O distance from 3.18 to 3.15 . However, the number of water hydrogen atoms around Cl does not change significantly as 6.0  6.7 with shortening Cl-D distance from 2.22 to 2.18 on compression. The pressure effect on the solvent water structure is also drastic as an increase in water oxygen atoms of 4.7 at the O-O distance of 2.79 at 0.1 MPa to 10.3 at 2.85 at 1 GPa. The number of water hydrogen atoms, however, does not change as 1.2 at the O-D distance of 1.74 for both pressures, demonstrating the presence of the O
6.7 with shortening Cl-D distance from 2.22 to 2.18 on compression. The pressure effect on the solvent water structure is also drastic as an increase in water oxygen atoms of 4.7 at the O-O distance of 2.79 at 0.1 MPa to 10.3 at 2.85 at 1 GPa. The number of water hydrogen atoms, however, does not change as 1.2 at the O-D distance of 1.74 for both pressures, demonstrating the presence of the O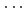 D hydrogen bonds which are significantly bent at 1 GPa at 298 K. This change of hydrogen bonds in water with pressure probably causes the drastic increase in water oxygen atoms around Cl.
D hydrogen bonds which are significantly bent at 1 GPa at 298 K. This change of hydrogen bonds in water with pressure probably causes the drastic increase in water oxygen atoms around Cl.
Kaneko, Masashi; Suzuki, Hideya; Matsumura, Tatsuro
Inorganic Chemistry, 57(23), p.14513 - 14523, 2018/12
Times Cited Count:24 Percentile:79.73(Chemistry, Inorganic & Nuclear)We elucidated the separation mechanism between Am(III) and Cm(III) ions by using two different types of diamide ligands, diglycolamide (DGA) and alkylated diamide amine (ADAAM), by means of the density functional theory technique and electron density analysis. The molecular geometries and formation reactions of the metal-ligand complexes were modeled by using [M(DGA)] and [M(ADAAM)(NO)(HO)]. We successfully reproduced Cm(III) selectivity over Am(III) with DGA and Am(III) selectivity over Cm(III) with ADAAM. Furthermore, we analyzed the bonding properties between the metal ion and the diamide-type ligands by using model complexes, [M(DGA)] and [M(ADAAM)(NO)(HO)], and revealed the differences in terms of the bond dissociation energy and the metal 5f-orbital participation in the covalency between the Am(III) and the Cm(III) complexes. It was suggested that the differences were key factors to understand the Am(III)/Cm(III) selectivity.
