


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Kai, Takeshi; Toigawa, Tomohiro; Matsuya, Yusuke*; Hirata, Yuho; Tsuchida, Hidetsugu*; Yokoya, Akinari*
Journal of Chemical Physics, 162(15), p.154102_1 - 154102_11, 2025/04
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Scientific knowledge of low-energy electrons resulting from water radiolysis is required to estimate radiation DNA damage. However, since the analysis of water radiolysis is very complex, this study focuses on the experimental values of low-energy electrons related to simple water photolysis and those generated by photoirradiation of electrodes in water. Both experimental analyses involve the presence or absence of a Coulomb field in the parent ion. In this study, we analyzed these experimental values using a calculation code that combines Monte Carlo and molecular dynamics methods. As a result, it was shown that the code reproduced the experimental values even under different experimental conditions, and the code was validated. The calculation code will be a powerful tool for analyzing the interaction between low-energy electrons and DNA, and is expected to be applied to elucidate the formation mechanism of radiation DNA damage.
 and machine learning potentials for modeling nuclear quantum effects in water
and machine learning potentials for modeling nuclear quantum effects in waterThomsen, B.; Nagai, Yuki*; Kobayashi, Keita; Hamada, Ikutaro*; Shiga, Motoyuki
Journal of Chemical Physics, 161(20), p.204109_1 - 204109_18, 2024/11
Times Cited Count:1 Percentile:37.13(Chemistry, Physical)We introduce the self-learning path integral hybrid Monte Carlo with mixed and machine learning potentials (SL-PIHMC-MIX) method which allows the application of hybrid Monte Carlo for both path integrals and for larger system sizes. The method shows savings of an order of magnitude with respect to the number of DFT calculations needed to calculate and converge the structure of room temperature water when using SL-PIHMC-MIX over ab initio path integral molecular dynamics (PIMD).
Tsuchida, Hidetsugu*; Tezuka, Tomoya*; Kai, Takeshi; Matsuya, Yusuke*; Majima, Takuya*; Saito, Manabu*
Journal of Chemical Physics, 161(10), p.104503_1 - 104503_8, 2024/09
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Although fast ion beams can damage DNA by chemical products such as secondary electrons produced by their interaction with water in living cells, the process of formation of these chemical products in the Bragg peak region used in particle therapy is not fully understood. To investigate this process, we performed experiments to evaluate the yields of radiolytic products produced when a liquid water jet in vacuum is irradiated with a MeV-energy carbon beam. In addition, ionization processes in water due to incident ions and secondary electrons were simulated using a radiation transport Monte Carlo code. The results indicated that the primary source of ionization in water is secondary electrons. Finally, we show that these elementary processes contribute to the development of radiation biophysics and biochemistry to study the formation mechanism of DNA damage.
Toigawa, Tomohiro; Kai, Takeshi; Kumagai, Yuta; Yokoya, Akinari*
Journal of Chemical Physics, 160(21), p.214119_1 - 214119_9, 2024/06
Times Cited Count:3 Percentile:75.15(Chemistry, Physical)The spur reaction is crucial for determining radiolysis or photolysis in liquid, but the spur expansion process has yet to be elucidated. One reason is the need to understand the role of the dielectric response of the solvating molecules surrounding the charged species generated by ionization. The dielectric response corresponds to the time evolution of the permittivity and might affect the chemical reaction-diffusion of the species in a spur expansion process. This study examined the competitive relationship between reaction-diffusion kinetics and the dielectric response by solving the Debye-Smoluchowski equation while considering the dielectric response. The Coulomb force between the charged species gradually decreases with the dielectric response. Our calculation results found a condition where fast recombination occurs before the dielectric response is complete. Although it has been reported that the primary G-values of free electrons depend on the static dielectric constant under low-linear-energy transfer radiation-induced ionization, we propose that considering the dielectric response can provide a deeper insight into fast recombination reactions under high-linear-energy transfer radiation- or photo-induced ionization. Our simulation method enables the understanding of fast radiation-induced phenomena in liquids.
 He spin filter and small-angle X-ray scattering
He spin filter and small-angle X-ray scatteringRyoki, Akiyuki*; Watanabe, Fumi*; Okudaira, Takuya*; Takahashi, Shingo*; Oku, Takayuki; Hiroi, Kosuke; Motokawa, Ryuhei; Nakamura, Yo*
Journal of Chemical Physics, 160(11), p.114907_1 - 114907_9, 2024/03
Times Cited Count:2 Percentile:37.13(Chemistry, Physical)Koshida, Hiroyuki*; Wilde, M.*; Fukutani, Katsuyuki
Journal of Chemical Physics, 160(3), p.034703_1 - 034703_9, 2024/01
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)Kai, Takeshi; Toigawa, Tomohiro; Ukai, Masatoshi*; Fujii, Kentaro*; Watanabe, Ritsuko*; Yokoya, Akinari*
Journal of Chemical Physics, 158(16), p.164103_1 - 164103_8, 2023/04
Times Cited Count:6 Percentile:69.12(Chemistry, Physical)New insight into water radiolysis and photolysis is indispensable in the dramatic progress of sciences and technologies in various research areas. In the radiation field, reactive hydrated electrons are considerably produced along radiation tracks. Although the formation results from a transient dynamic correlation between ejected electrons and water, the individual mechanisms of electron thermalization, delocalization, and polarization are unknown. Using a dynamic Monte Carlo code, we show herein that the ejected electrons are immediately delocalized by molecular excitations in parallel with phonon polarization and gradually thermalized by momentum transfer with an orientation polarization in a simultaneous manner. Our results show that these mechanisms heavily depend on the intermolecular vibration and rotation modes peculiar to water. We expect our approach to be a powerful technique for connecting physical and chemical processes in various solvents.
 species at SiO/Si interfaces in Si dry oxidation; Comparison between p-Si(001) and n-Si(001) surfaces
species at SiO/Si interfaces in Si dry oxidation; Comparison between p-Si(001) and n-Si(001) surfacesTsuda, Yasutaka; Yoshigoe, Akitaka; Ogawa, Shuichi*; Sakamoto, Tetsuya*; Yamamoto, Yoshiki*; Yamamoto, Yukio*; Takakuwa, Yuji*
Journal of Chemical Physics, 157(23), p.234705_1 - 234705_21, 2022/12
Times Cited Count:2 Percentile:14.89(Chemistry, Physical)Akazawa, Daisuke; Sasaki, Takehiko*; Nagasaka, Masanari*; Shiga, Motoyuki
Journal of Chemical Physics, 156(4), p.044202_1 - 044202_7, 2022/01
Times Cited Count:8 Percentile:62.23(Chemistry, Physical)The hydration structure of cellulose is very important for understanding the hydrolysis of cellulose at the molecular level. In this paper, we report a joint experimental and theoretical study on the X-ray absorption spectroscopy (XAS) of aqueous cellobiose, a disaccharide unit of cellulose. In the experimental part, high resolution measurements of the carbon K-edge XAS spectra were performed. It was found that the peak heights in the spectrum change considerably over the temperature range of 25  C to 60 C, which is a reflection of the number of hydrogen bonds between cellobiose and water. We suggest that this spectral change could be useful information for identifying the hydration of cellulose in various environments.
C to 60 C, which is a reflection of the number of hydrogen bonds between cellobiose and water. We suggest that this spectral change could be useful information for identifying the hydration of cellulose in various environments.
Nagaya, Yuki*; Nakatsu, Hiroki*; Ogura, Shohei*; Shimazaki, Kota*; Ueta, Hirokazu; Takeyasu, Kotaro*; Fukutani, Katsuyuki
Journal of Chemical Physics, 155(19), p.194201_1 - 194201_6, 2021/11
Times Cited Count:1 Percentile:4.26(Chemistry, Physical)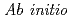 study of nuclear quantum effects on sub- and supercritical water
study of nuclear quantum effects on sub- and supercritical waterThomsen, B.; Shiga, Motoyuki
Journal of Chemical Physics, 155(19), p.194107_1 - 194107_11, 2021/11
Times Cited Count:8 Percentile:47.53(Chemistry, Physical)Kobayashi, Keita; Nagai, Yuki; Itakura, Mitsuhiro; Shiga, Motoyuki
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
Times Cited Count:13 Percentile:67.66(Chemistry, Physical)no abstracts in English
path integral molecular dynamicsThomsen, B.; Shiga, Motoyuki
Journal of Chemical Physics, 154(8), p.084117_1 - 084117_10, 2021/02
Times Cited Count:14 Percentile:70.46(Chemistry, Physical)In this study we investigate the nuclear quantum effects on the acidity constant of liquid water isotopologues at the ambient condition by path integral molecular dynamics simulations. This technique not only reproduces the acidity constants of liquid DO experimentally measured but also allows for a theoretical prediction of the acidity constants of liquid TO, aqueous HDO and HTO, which are unknown due to its scarcity. The results indicate that the nuclear quantum effects play an indispensable role in the absolute determination of acidity constants.
Noguchi, Yoshifumi*; Hiyama, Miyabi*; Shiga, Motoyuki; Akiyama, Hidefumi*; Sugino, Osamu*
Journal of Chemical Physics, 153(20), p.201103_1 - 201103_6, 2020/11
Times Cited Count:2 Percentile:8.04(Chemistry, Physical)Stabilizing mechanisms of three isomers of the aqueous oxyluciferin in the first excited state were investigated using first-principles molecular dynamics simulations. Only the phenolate-keto isomer became attracted to the water molecules in its excited state and was stabilized by increasing the number of hydrogen bonds with nearby water molecules. The most stable isomer in the excited state was the phenolate-keto, and the phenolate-enol and phenol-enolate isomers were higher in energy by 0.38 eV and 0.57 eV, respectively, than the phenolate-keto. This was in contrast to the case of ground state in which the phenolate-enol was the most stable isomer.
Akahama, Yuichi*; Miyakawa, Masashi*; Taniguchi, Takashi*; Sano, Asami; Machida, Shinichi*; Hattori, Takanori
Journal of Chemical Physics, 153(1), p.014704_1 - 014704_5, 2020/07
Times Cited Count:11 Percentile:48.91(Chemistry, Physical)The structure refinement of black phosphorus was performed at pressures of up to 3.2 GPa at room temperature by powder neutron diffraction techniques. The bond lengths and bond angles between the phosphorus atoms at pressures were precisely determined and confirmed to be consistent with those of the previous single crystal X-ray analysis [Brown and Randqvist, Acta Cryst. 19, 684 (1965)]. Although lattice parameters exhibited an anisotropic compressibility, the covalent P1-P2 and P1-P3 bond lengths were almost independent of pressure and only the P3-P1-P2 bond angle was reduced significantly. On the basis of our results, the significant discrepancy in the bond length reported by Cartz et al. [J. Chem. Phys. 71, 1718 (1979)] has been solved. Our structural data will contribute to the elucidation of the Dirac semimetal state of black phosphorus under high pressure.
 O
OYamakawa, Koichiro; Nasu, Hirokazu*; Suzuki, Natsumi*; Shimizu, Genki*; Arakawa, Ichiro*
Journal of Chemical Physics, 152(17), p.174310_1 - 174310_13, 2020/05
Times Cited Count:2 Percentile:8.04(Chemistry, Physical)We have established an apparatus for terahertz and mid-infrared spectroscopy in an ultrahigh vacuum and have measured absorption spectra of DO clusters trapped in solid Ar. To assign terahertz absorption peaks due to the DO dimer, trimer, and tetramer, the dependence of the spectrum on the annealing temperature and DO dilution was analyzed. The assignment was also examined by ab initio calculations with use of the ONIOM method, where flexibility of surrounding Ar atoms was systematically incorporated. We identified all of the intermolecular fundamentals of the dimer and those with significant intensities of the trimer and tetramer, whose structural symmetries were revealed to be broken down. After isolating the DO clusters in solid Ar, we sublimated only Ar atoms to leave behind matrix-sublimation ice, which was found to be amorphous- or crystal-like depending on the formation conditions: the dilution and sublimation-temperature. Since the crystallinity got higher with raising the dilution and sublimation-temperature, the diffusion of the DO monomer on the surface of sublimating solid Ar was found to be crucial to the crystallization of the sublimation ice.
 irradiation
irradiationOkazaki, Hiroyuki*; Kakitani, Kenta*; Kimata, Tetsuya*; Idesaki, Akira*; Koshikawa, Hiroshi*; Matsumura, Daiju; Yamamoto, Shunya*; Yamaki, Tetsuya*
Journal of Chemical Physics, 152(12), p.124708_1 - 124708_5, 2020/03
Times Cited Count:5 Percentile:24.42(Chemistry, Physical)(101) surfaceNagatsuka, Naoki*; Wilde, M.*; Fukutani, Katsuyuki
Journal of Chemical Physics, 152(7), p.074708_1 - 074708_6, 2020/02
Times Cited Count:13 Percentile:59.54(Chemistry, Physical)Sugimoto, Takeru*; Nasu, Hirokazu*; Arakawa, Ichiro*; Yamakawa, Koichiro
Journal of Chemical Physics, 150(18), p.184302_1 - 184302_5, 2019/05
Times Cited Count:4 Percentile:17.43(Chemistry, Physical)We measured infrared absorption spectra of crystalline II of methane and succeeded in detecting a prominent Q(2) peak by rapid cooling after annealing as well as previously reported rovibrational and librational-vibrational peaks. The integral intensities of the R(0), R(1), and Q(2) peaks were found to show biexponential dependence on time. This clearly demonstrates the interconversion among the three nuclear-spin isomers occupying low-lying rotational levels.
Yoshida, Koji*; Inoue, Takuya*; Torigoe, Motokatsu*; Yamada, Takeshi*; Shibata, Kaoru; Yamaguchi, Toshio*
Journal of Chemical Physics, 149(12), p.124502_1 - 124502_10, 2018/09
Times Cited Count:4 Percentile:14.97(Chemistry, Physical)Differential scanning calorimetry, X-ray diffraction, and quasi-elastic neutron scattering (QENS) measurements of aqueous glycine solutions confined in mesoporous silica (MCM-41) were performed at different glycine concentrations, pH, and loading ratio (= mass of glycine solution / mass of dry MCM-41) in the temperature range from 305 to 180 K to discuss the confinement effect on the thermal behavior, the structure, and the dynamic properties of the solutions.
