


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Kido, Kentaro; Kaneko, Masashi
Journal of Computational Chemistry, 44(4), p.546 - 558, 2023/02
Times Cited Count:1 Percentile:5.83(Chemistry, Multidisciplinary)Shiga, Motoyuki
Journal of Computational Chemistry, 43(27), p.1864 - 1879, 2022/10
Times Cited Count:7 Percentile:47.13(Chemistry, Multidisciplinary)A new approximate method for quantum vibrational dynamics (Brownian chain molecular dynamics: BCMD) based on the path integral method is proposed. Conventional methods such as centroid molecular dynamics and ring polymer dynamics cannot correctly calculate quantum vibration spectra under low temperature conditions. Thus, I introduce an over-damped Langevin equation for the non-centroid degrees of freedom of atoms to suppress unphysical resonances and vibrational rotational couplings, which are the source of the problem. I verify this approximation through applications to infrared absorption spectra of light and heavy water. I also demonstrate the feasibility of ab initio BCMD simulations by the combination with electronic structure theory.
Pal, A.*; Pal, S.*; Verma, S.*; Shiga, Motoyuki; Nair, N. N.*
Journal of Computational Chemistry, 42(28), p.1996 - 2003, 2021/10
Times Cited Count:11 Percentile:51.41(Chemistry, Multidisciplinary)Temperature-accelerated sliced sampling is a computational method for high-dimensional free energy landscapes. In the conventional method, post-processing of the calculation has been required using the weighted histogram analysis method. In this study, we establish a refined approach without post-processing using the average force. We demonstrated the cases of the two-dimensional and four-dimensional free energy landscapes of alanine dipeptide and tripeptide, where they were computed within an error of about kcal/mol. Vast application of this method is expected for free energy calculations in computational chemistry.
Kondo, Tomomi; Sasaki, Takehiko*; Shiga, Motoyuki
Journal of Computational Chemistry, 42(25), p.1783 - 1791, 2021/09
Times Cited Count:5 Percentile:24.90(Chemistry, Multidisciplinary)Sugar alcohol dehydration in hot water is an important reaction that allows for environmentally friendly biomass conversion without the use of organic solvents. Here, we report a free-energy analysis by metadynamics (MTD) simulations based on ab initio and semiempirical electronic structure theory to understand the mechanism of dehydration reactions of sorbitol (SBT) in hot acidic water. It was found that the reaction proceeds via an S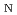 2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected by the acidic species. The free energy barriers of the reaction pathways leading to five-membered ether products are lower than those leading to six-membered ether products.
2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected by the acidic species. The free energy barriers of the reaction pathways leading to five-membered ether products are lower than those leading to six-membered ether products.
Kondo, Tomomi; Sasaki, Takehiko*; Ruiz-Barragan, S.*; Ribas-Ari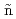 o, J.*; Shiga, Motoyuki; Ruiz-Barragan, S.*
o, J.*; Shiga, Motoyuki; Ruiz-Barragan, S.*
Journal of Computational Chemistry, 42(3), p.156 - 165, 2021/01
Times Cited Count:5 Percentile:19.73(Chemistry, Multidisciplinary)We propose a canonical sampling method to refine metadynamics simulations a posteriori. This approach could be useful particularly when two or more free energy barriers are to be compared among chemical reactions in different or competing conditions. The method was then applied to study the acid dependence of polyalcohol dehydration reactions in high-temperature aqueous solutions. It was found that the reaction proceeds consistently via an S2 mechanism, whereby the free energy of protonation of the hydroxyl group created as an intermediate is affected significantly by the acidic species.
Kido, Kentaro
Journal of Computational Chemistry, 40(24), p.2072 - 2085, 2019/09
Times Cited Count:3 Percentile:10.79(Chemistry, Multidisciplinary)Sakuraba, Shun; Matsubayashi, Nobuyuki*
Journal of Computational Chemistry, 35(21), p.1592 - 1608, 2014/08
Times Cited Count:61 Percentile:80.66(Chemistry, Multidisciplinary)ERmod is a software package to efficiently and approximately compute the solvation free energy using the method of energy representation. Molecular simulation is to be conducted at two condensed-phase systems of the solution of interest and the reference solvent with test-particle insertion of the solute. The subprogram 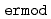 in ERmod then provides a set of energy distribution functions from the simulation trajectories, and another subprogram
in ERmod then provides a set of energy distribution functions from the simulation trajectories, and another subprogram 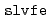 determines the solvation free energy from the distribution functions through an approximate functional. The present paper describes the design and implementation of ERmod, and illustrates its performance in solvent water for two organic solutes and two protein solutes. Actually, the free-energy computation with ERmod is not restricted to the solvation in homogeneous medium such as fluid and polymer, and can treat the binding into weakly ordered system with nano-inhomogeneity such as micelle and lipid membrane. ERmod is available on web at
determines the solvation free energy from the distribution functions through an approximate functional. The present paper describes the design and implementation of ERmod, and illustrates its performance in solvent water for two organic solutes and two protein solutes. Actually, the free-energy computation with ERmod is not restricted to the solvation in homogeneous medium such as fluid and polymer, and can treat the binding into weakly ordered system with nano-inhomogeneity such as micelle and lipid membrane. ERmod is available on web at  .
.
Ikebe, Kimiyoshi; Sakuraba, Shun; Kono, Hidetoshi
Journal of Computational Chemistry, 35(1), p.39 - 50, 2014/01
Times Cited Count:17 Percentile:44.03(Chemistry, Multidisciplinary)Fujimoto, Hirofumi*; Higuchi, Mariko; Koike, Manabu*; Ode, Hirotaka*; Pinak, M.; Kotulic Bunta, J.*; Nemoto, Toshiyuki*; Sakudo, Takashi*; Honda, Naoko*; Maekawa, Hideaki*; et al.
Journal of Computational Chemistry, 33(3), p.239 - 246, 2012/01
Times Cited Count:36 Percentile:66.42(Chemistry, Multidisciplinary)Lysine acetylation is one of the most common protein post transcriptional modifications. The acetylation effects of lysine residues on Ku protein were examined herein applying several computer simulation techniques. Acetylation of the lysine residues did not reduce the affinity between Ku and its substrate, DNA, in spite of the fact that the substitution of lysine with glutamine (KQ mutant) reduced the affinity of Ku for DNA, or the substitution of lysine with arginine (KR mutant) did not reduce it, as previously reported in experimental studies. These results suggest that the effects of in vivo acetylation may be overestimated when the KQ mutant is employed in mimicry of the acetylated protein.
Ishida, Hisashi
Journal of Computational Chemistry, 31(12), p.2317 - 2329, 2010/09
Times Cited Count:9 Percentile:34.09(Chemistry, Multidisciplinary)Fujimoto, Hirofumi*; Pinak, M.; Nemoto, Toshiyuki*; O'Neill, P.*; Kume, Etsuo; Saito, Kimiaki; Maekawa, Hideaki*
Journal of Computational Chemistry, 26(8), p.788 - 798, 2005/06
Times Cited Count:24 Percentile:57.93(Chemistry, Multidisciplinary)Clustered DNA damage sites induced by ionizing radiation have been suggested to have serious consequences to organisms. In this study, approaches based on molecular dynamics (MD) simulation have been applied to examine conformational changes and energetic properties of DNA molecules containing clustered damage sites consisting of 2 lesioned sites, 8-oxoG and AP site. After 1 nanosecond of MD simulation, one of the 6 DNA molecules containing a clustered damage site develop specific characteristic features: sharp bending at the lesioned site and weakening or complete loss of electrostatic interaction energy between 8-oxoG and bases locating on the complementary strand. From these results it is suggested that these changes would make it difficult for the repair enzyme to bind to the lesions within the clustered damage site and thereby result in a reduction of its repair capacity.
Pinak, M.
Journal of Computational Chemistry, 24(7), p.898 - 907, 2003/04
Times Cited Count:8 Percentile:35.73(Chemistry, Multidisciplinary)The molecular dynamics (MD) simulation of DNA mutagenic oxidative lesion 8-oxoG, complexed with the repair enzyme - hOGG1 was performed in order to describe the dynamical process of DNA-enzyme complex formation. After 500 picoseconds of MD the lesioned DNA and enzyme formed a complex that lasted stable until the simulation was terminated at 1 ns. The N-terminus of arginine 313 was located close to the phosphodiester bond of nucleotide with 8-oxoG enabling chemical reactions between amino acid and lesion. Phosphodiester bond at C5' of 8-oxoG was displaced to the position close to the N-terminus of arginine 313. The water mediated hydrogen bonds network was formed in each contact area between DNA and enzyme further enhancing the stability of complex.
Pinak, M.
Journal of Computational Chemistry, 22(15), p.1723 - 1731, 2001/11
Times Cited Count:3 Percentile:21.81(Chemistry, Multidisciplinary)no abstracts in English

