


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
He, X.*; Kagi, Hiroyuki*; Komatsu, Kazuki*; Iizuka, Riko*; Okajima, Hajime*; Hattori, Takanori; Sano, Asami; Machida, Shinichi*; Abe, Jun*; Goto, Hirotada*; et al.
Journal of Molecular Structure, 1310, p.138271_1 - 138271_8, 2024/08
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)High-pressure responses of the O-D F hydrogen bonds in deuterated magnesium hydroxyfluoride were investigated using neutron powder diffraction and Raman spectroscopy. The Rietveld analysis at ambient conditions revealed a chemical formula of Mg(OD)
F hydrogen bonds in deuterated magnesium hydroxyfluoride were investigated using neutron powder diffraction and Raman spectroscopy. The Rietveld analysis at ambient conditions revealed a chemical formula of Mg(OD)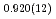 F
F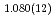 and hydroxyl group/fluorine disorder (OD/F disorder) in the crystal structure, which gave rise to two hydrogen-bonding configurations. The Rietveld analysis showed the hydrogen-bonding geometries remains up to 9.8 GPa, indicating no pressure-induced strengthening of hydrogen bonds. The Raman spectra at ambient conditions showed three hydroxyl stretching bands at 2613, 2694, and 2718 cm
and hydroxyl group/fluorine disorder (OD/F disorder) in the crystal structure, which gave rise to two hydrogen-bonding configurations. The Rietveld analysis showed the hydrogen-bonding geometries remains up to 9.8 GPa, indicating no pressure-induced strengthening of hydrogen bonds. The Raman spectra at ambient conditions showed three hydroxyl stretching bands at 2613, 2694, and 2718 cm . The high frequencies of the O-D stretching modes indicated that the hydroxyls should be involved in weak or none hydrogen-bonding interactions. Up to 20.2 GPa, the mode initially centered at 2694 cm displayed a pressure-induced blue shift, revealing no strengthening of hydrogen bonds under compression. We discuss the existence of hydrogen bonds and the causes of the blue-shifting hydroxyls at ambient and at high pressures.
. The high frequencies of the O-D stretching modes indicated that the hydroxyls should be involved in weak or none hydrogen-bonding interactions. Up to 20.2 GPa, the mode initially centered at 2694 cm displayed a pressure-induced blue shift, revealing no strengthening of hydrogen bonds under compression. We discuss the existence of hydrogen bonds and the causes of the blue-shifting hydroxyls at ambient and at high pressures.
Tabata, Chihiro; Watanabe, Hirohito*; Shirasaki, Kenji*; Sunaga, Ayaki*; Fukuda, Takamitsu*; Li, D.*; Yamamura, Tomoo*
Journal of Molecular Structure, 1277, p.134870_1 - 134870_8, 2023/04
Times Cited Count:4 Percentile:20.92(Chemistry, Physical)Neutral and cationic U(IV) sandwiched phthalocyanine (Pc) complexes were prepared. The neutral species, UPc , was obtained by the reaction of UCl
, was obtained by the reaction of UCl ] and phthalonitrile, and the [UPc][BF] crystals were grown by electrolysis. The structures of the complexes were determined crystallographically. A U(IV) ion has two
] and phthalonitrile, and the [UPc][BF] crystals were grown by electrolysis. The structures of the complexes were determined crystallographically. A U(IV) ion has two 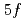 electrons, which carry a magnetic moment localized at the U sites, making the complex magnetically active. The magnetic susceptibility measurement of UPc revealed that the system was paramagnetic with local magnetic moments down to 2 K, but these magnetic moments were suppressed, possibly by a ligand field effect. The paramagnetism was also confirmed by the magnetization curve without hysteresis. The cationic complex, [UPc][BF]] crystalized in a tetragonal structure with the space group
electrons, which carry a magnetic moment localized at the U sites, making the complex magnetically active. The magnetic susceptibility measurement of UPc revealed that the system was paramagnetic with local magnetic moments down to 2 K, but these magnetic moments were suppressed, possibly by a ligand field effect. The paramagnetism was also confirmed by the magnetization curve without hysteresis. The cationic complex, [UPc][BF]] crystalized in a tetragonal structure with the space group 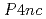 , in which the UPc molecules stacked along the tetragonal
, in which the UPc molecules stacked along the tetragonal 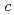 -axis. The structure was similar to that of [LnPc][BF]], but with distinct disorder in the stacking plane. The stabilities of the crystal and molecular structures and the electronic configurations of UPc and [UPc][BF]] were evaluated via the
-axis. The structure was similar to that of [LnPc][BF]], but with distinct disorder in the stacking plane. The stabilities of the crystal and molecular structures and the electronic configurations of UPc and [UPc][BF]] were evaluated via the 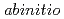 calculations that included the multiconfigurational nature of the actinide element.
calculations that included the multiconfigurational nature of the actinide element.
Arakawa, Masashi*; Kagi, Hiroyuki; Fukazawa, Hiroshi
Journal of Molecular Structure, 972(1-3), p.111 - 114, 2010/05
Times Cited Count:7 Percentile:15.44(Chemistry, Physical)We measured the neutron powder diffraction of 0.013 M KOD-doped DO ice to investigate the formation process of ice XI, a hydrogen-ordered phase of ice Ih. The doped ice Ih transformed to ice XI after annealing at 57 and subsequently at 68 K. The mass fraction of ice XI to that of the doped ice (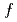 ) was estimated using Rietveld analysis for each sample. The value of the doped ice, which had once experienced being ice XI ( = 0.23), was larger than that of the doped ice, which had never experienced being ice XI ( = 0.14). Results indicate that small hydrogen-ordered domains remained in the ice Ih, which had once trans- formed to ice XI, and accelerated the phase transition from ice Ih to ice XI. Results further suggest that large amounts of ice on icy bodies in our solar system can transform to ice XI, which might be detectable using infrared telescopes or planetary exploration in the near future.
) was estimated using Rietveld analysis for each sample. The value of the doped ice, which had once experienced being ice XI ( = 0.23), was larger than that of the doped ice, which had never experienced being ice XI ( = 0.14). Results indicate that small hydrogen-ordered domains remained in the ice Ih, which had once trans- formed to ice XI, and accelerated the phase transition from ice Ih to ice XI. Results further suggest that large amounts of ice on icy bodies in our solar system can transform to ice XI, which might be detectable using infrared telescopes or planetary exploration in the near future.
Kurosaki, Yuzuru; Yokoyama, Keiichi; Yokoyama, Atsushi
Journal of Molecular Structure; THEOCHEM, 913(1-3), p.38 - 42, 2009/11
 SO
SO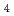
 (HO)
(HO)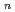 (
(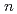 =1-6) on semiempirical PM6 potential surfaces
=1-6) on semiempirical PM6 potential surfacesKakizaki, Akira*; Motegi, Haruki*; Yoshikawa, Takehiro*; Takayanagi, Toshiyuki*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 901(1-3), p.1 - 8, 2009/05
Quantum path-integral molecular dynamics simulations for a series of sulfuric acid clusters, HSO(HO)(=1-6), were performed using semiempirical PM6 method. It was found that the acid dissociation probability increases with an increase in the cluster size, and that so-called contact-ion-pair structures are dominant in the proton-dissociated clusters. The importance of nuclear quantum effects in the cluster structures and proton-transfer processes is demonstrated.
O)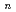 (=2-7) clusters on semiempirical PM6 potential energy surfaces
(=2-7) clusters on semiempirical PM6 potential energy surfacesTakayanagi, Toshiyuki*; Yoshikawa, Takehiro*; Kakizaki, Akira*; Shiga, Motoyuki; Tachikawa, Masanori*
Journal of Molecular Structure; THEOCHEM, 869(1-3), p.29 - 36, 2008/11
Molecular dynamics simulations based on semiempirical PM6 method was carried out to study statistical structures of glycine-water clusters. We found that zwitterionic form is more stable than neutral form as the cluster size increases. The difference in water solvation structures around the neutral or zwitterionic glycine form is also discussed.
Kurosaki, Yuzuru
Journal of Molecular Structure; THEOCHEM, 850(1-3), p.9 - 16, 2008/02
Times Cited Count:12 Percentile:29.10(Chemistry, Physical)Direct density-functional (DFT) molecular-dynamics (MD) calculations have been carried out for the following two hydrogen atom production channels in acetaldehyde photodissociation on the lowest triplet-state (T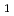 ) potential energy surface (PES): CD
) potential energy surface (PES): CD CHO
CHO CDCHO+D(1); CDCHOCDCO+H(2). The employed DFT method was B3LYP with the cc-pVDZ basis set. The average product hydrogen kinetic energies estimated from the results of the direct DFT MD calculations were 18.3 and 16.6 kcal mol for reactions 1 and 2, respectively, and these were half - two thirds of the previously measured values [Kang et al. Chem. Phys. Lett. 434 (2007) 6]. This is because of the low reverse barrier heights predicted at the B3LYP/cc-pVDZ level. The present results for the product hydrogen kinetic energies, however, agree qualitatively with the experimental measurements and strongly supports the mechanisms taking place on the T PES.
CDCHO+D(1); CDCHOCDCO+H(2). The employed DFT method was B3LYP with the cc-pVDZ basis set. The average product hydrogen kinetic energies estimated from the results of the direct DFT MD calculations were 18.3 and 16.6 kcal mol for reactions 1 and 2, respectively, and these were half - two thirds of the previously measured values [Kang et al. Chem. Phys. Lett. 434 (2007) 6]. This is because of the low reverse barrier heights predicted at the B3LYP/cc-pVDZ level. The present results for the product hydrogen kinetic energies, however, agree qualitatively with the experimental measurements and strongly supports the mechanisms taking place on the T PES.
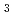 in LiCl-KCl eutectic melt
in LiCl-KCl eutectic meltOkamoto, Yoshihiro; Yaita, Tsuyoshi; Minato, Kazuo
Journal of Molecular Structure, 749(1-3), p.70 - 73, 2005/07
Times Cited Count:3 Percentile:7.17(Chemistry, Physical)The local structures of molten BiCl and its mixtures in LiCl-KCl eutectic melt were investigated by X-ray absorption fine structure (XAFS) technique. The 1st Bi-Cl correlation in molten pure BiCl shows covalent nature, since the distance was almost the same as sum of covalent radii of Bi and Cl and the coordination number was almost 3. The similar property was observed also in the mixture of 75% BiCl with LiCl-KCl eutectic melt. Drastic change was detected in 25% BiCl mixture melt. The 1st Bi-Cl distance was sum of ionic radii in molten 25% BiCl melt. The results suggest that BiCl changes from molecular liquid to ionic by mixing with alkali chlorides.
Fujimoto, Hirofumi; Pinak, M.; Nemoto, Toshiyuki*; Sakamoto, Kiyotaka*; Yamada, Kazuyuki*; Hoshi, Yoshiyuki*; Kume, Etsuo
Journal of Molecular Structure; THEOCHEM, 681(1-3), p.1 - 8, 2004/01
We developed the novel system, Fujitsu Bio Molecular Visualization System (F-BMVS), that enables to produce real pictures and an animation by arranging them along a time series of a large scale simulation of biomolecules associated with a molecular dynamics (MD) simulation program. This animation system is used to study the results of molecular dynamics code, AMBER, in order to find structural differences on the lesioned DNA comparing with non-damaged DNA. These structural differences would be a factor that guides a repair enzyme to discriminate a lesion from non-damaged DNA region.
by K-absorption edge XAFSOkamoto, Yoshihiro; Shiwaku, Hideaki; Yaita, Tsuyoshi; Narita, Hirokazu; Tanida, Hajime*
Journal of Molecular Structure, 641(1), p.71 - 76, 2002/10
Times Cited Count:43 Percentile:71.88(Chemistry, Physical)The local structure of molten LaCl was investigated by X-ray absorption fine structure(XAFS) of the La K-edge. The nearest La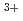 -Cl
-Cl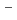 distance andcoordination number were 2.89
distance andcoordination number were 2.89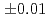
 and 7.4
and 7.4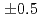 from the curve fitting of the 1st peak in the fourier transform magnitude
from the curve fitting of the 1st peak in the fourier transform magnitude 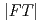 . The coordination number larger than 6 suggests that the local structure of molten LaCl is not a simple octahedral coordination (LaCl
. The coordination number larger than 6 suggests that the local structure of molten LaCl is not a simple octahedral coordination (LaCl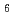 )
)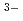 , but 7-fold (LaCl
, but 7-fold (LaCl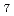 )
)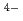 and/or 8-fold (LaCl
and/or 8-fold (LaCl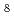 )
)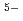 complexes. The 1st La-La distance, of which correlation was observed as a weak 2nd peak in the , was evaluated to be 4.9. It suggests that the distorted corner-sharing connection of the complex species is predominant in the melt, inontrast with molten YCl in which the edge-sharing connection of the 6-fold (YCl) mainly exists.
complexes. The 1st La-La distance, of which correlation was observed as a weak 2nd peak in the , was evaluated to be 4.9. It suggests that the distorted corner-sharing connection of the complex species is predominant in the melt, inontrast with molten YCl in which the edge-sharing connection of the 6-fold (YCl) mainly exists.
Ishida, Hisashi
Journal of Biomolecular Structure and Dynamics, 19(5), p.839 - 851, 2002/05
Times Cited Count:11 Percentile:25.85(Biochemistry & Molecular Biology)no abstracts in English
Pinak, M.
Journal of Molecular Structure; THEOCHEM, 583(1-3), p.189 - 197, 2002/04
The local structural and energetic impact of a mutagenic oxidative lesion 7,8-dihydro-8-oxoguanine (8-oxoG) on a DNA molecule was studied by the method of a molecular dynamics (MD) simulation. The molecule of 8-oxoG was inserted into central part of B-DNA 15-mer d(GCGTCCA'8-oxoG'GTCTACC) replacing the native guanine. The 2-nanosecond MD simulations were performed with the AMBER 5.0 program code at the constant temperature of 310 K ( 36.5ºC, temperature of human body) for the 8-oxoG lesioned and native DNA molecules. The broken hydrogen bonds resulting in locally collapsed B-DNA structure were observed at the lesion site. The adenine 21 on the complementary strand (separated from 8-oxoG by 1 base pair) is flipped-out of the DNA double helix. Its extrahelical position forms a hole that may favor docking of repair enzyme into DNA during repair process. A strong electrostatic repulsion between nucleotide with the 8-oxoG and neighboring nucleotides contributes to the observed instability of DNA at the lesion.
36.5ºC, temperature of human body) for the 8-oxoG lesioned and native DNA molecules. The broken hydrogen bonds resulting in locally collapsed B-DNA structure were observed at the lesion site. The adenine 21 on the complementary strand (separated from 8-oxoG by 1 base pair) is flipped-out of the DNA double helix. Its extrahelical position forms a hole that may favor docking of repair enzyme into DNA during repair process. A strong electrostatic repulsion between nucleotide with the 8-oxoG and neighboring nucleotides contributes to the observed instability of DNA at the lesion.
 F
F (n=2-5) clusters
(n=2-5) clustersHaketa, Naoki*; Yokoyama, Keiichi; Tanaka, Hiromasa*; Kudo, Hiroshi*
Journal of Molecular Structure; THEOCHEM, 577(1), p.55 - 67, 2002/01
no abstracts in English
H + Cl CHCl + Cl reaction; Ab initio molecular orbital studyKurosaki, Yuzuru
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
The CASSCF and MRCI calculations with the cc-pVTZ basis set have been carried out for the CH + Cl CHCl + Cl reaction. It has been revealed that the reaction has a small barrier from the CHCl + Cl side at the CASSCF level of theory, but it has no barrier at the MRCI level. Namely, the CHCl + Cl CH + Cl reaction was predicted to be a spontaneous reaction. The result of the MRCI calculation strongly supports the prediction of our previous PMP4(SDTQ) calculation [J. Mol. Struct. (Theochem) 503 (2000) 231].
NO( B)NO(
B)NO(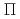 )+H reaction
)+H reactionKurosaki, Yuzuru*; Takayanagi, Toshiyuki
Journal of Molecular Structure; THEOCHEM, 507(1-3), p.119 - 126, 2000/07
no abstracts in English
H+ClCHCl reactionKurosaki, Yuzuru*
Journal of Molecular Structure; THEOCHEM, 503(3), p.231 - 240, 2000/05
no abstracts in English
Pinak, M.*
Journal of Molecular Structure; THEOCHEM, 499, p.57 - 70, 2000/03
no abstracts in English
D)+CH insertion reactionTakayanagi, Toshiyuki; Kurosaki, Yuzuru*
Journal of Molecular Structure; THEOCHEM, 492, p.151 - 158, 1999/00
no abstracts in English
Pinak, M.*
Journal of Molecular Structure; THEOCHEM, 466, p.219 - 234, 1999/00
no abstracts in English
elimination from 2,3-dimethylbutane cation: (CH)CHCH(CH)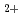 (CH)C=C(CH)+H; An ab initio molecular orbital study
(CH)C=C(CH)+H; An ab initio molecular orbital studyKurosaki, Yuzuru*; Takayanagi, Toshiyuki; Miyazaki, Tetsuro*
Journal of Molecular Structure; THEOCHEM, 452, p.209 - 218, 1998/00
no abstracts in English
