


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Shinohara, Yuya*; Iwashita, Takuya*; Nakanishi, Masahiro*; Osti, N. C.*; Kofu, Maiko; Nirei, Masami; Dmowski, W.*; Egami, Takeshi*
Journal of Physical Chemistry B, 128(6), p.1544 - 1549, 2024/02
Times Cited Count:2 Percentile:44.97(Chemistry, Physical)Massey, D.*; Williams, C. D.*; Mu, J.*; Masters, A. J.*; Motokawa, Ryuhei; Aoyagi, Noboru; Ueda, Yuki; Antonio, M. R.*
Journal of Physical Chemistry B, 127(9), p.2052 - 2065, 2023/03
Times Cited Count:2 Percentile:14.56(Chemistry, Physical)Kusaka, Ryoji; Watanabe, Masayuki
Journal of Physical Chemistry B, 125(24), p.6727 - 6731, 2021/06
Times Cited Count:13 Percentile:46.94(Chemistry, Physical)Chang, Y. L.*; Sasaki, Takehiko*; Ribas-Ari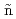 o, J.*; Machida, Masahiko; Shiga, Motoyuki
o, J.*; Machida, Masahiko; Shiga, Motoyuki
Journal of Physical Chemistry B, 123(7), p.1662 - 1671, 2019/02
Times Cited Count:4 Percentile:8.95(Chemistry, Physical)Dehydration of biomass-derived polyalcohols has recently drawn attention in green chemistry as a prototype of selective reactions controllable in hot water or hot carbonated water, without any use of organic solvents or metal catalysts. Here, we report a free-energy analysis based on first-principles metadynamics and blue-moon ensemble simulations to understand the mechanism of competing intramolecular dehydration reactions of 1,2,5-pentanetriol in hot acidic water. The simulations consistently predict that the most dominant mechanism is the proton-assisted S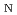 2 process, where the protonation of the hydroxyl group by water and the C-O bond breaking and formation occur in a single step. The detailed mechanism found from the simulations shows how the reaction paths are selective in hot water and why the reaction rates are accelerated in acidic environments, thus giving a clear explanation of experimental findings for a broad class of competing dehydration processes of polyalcohols.
2 process, where the protonation of the hydroxyl group by water and the C-O bond breaking and formation occur in a single step. The detailed mechanism found from the simulations shows how the reaction paths are selective in hot water and why the reaction rates are accelerated in acidic environments, thus giving a clear explanation of experimental findings for a broad class of competing dehydration processes of polyalcohols.
Mu, J.*; Motokawa, Ryuhei; Akutsu, Kazuhiro*; Nishitsuji, Shotaro*; Masters, A. J.*
Journal of Physical Chemistry B, 122(4), p.1439 - 1452, 2018/02
Times Cited Count:41 Percentile:71.84(Chemistry, Physical)Sekine, Yurina; Endo, Hitoshi*; Iwase, Hiroki*; Takeda, Shigeo*; Mukai, Sadaatsu*; Fukazawa, Hiroshi; Littrell, K. C.*; Sasaki, Yoshihiro*; Akiyoshi, Kazunari*
Journal of Physical Chemistry B, 120(46), p.11996 - 12002, 2016/11
Times Cited Count:12 Percentile:29.05(Chemistry, Physical)The detailed structure of a nanogel formed by self-association of cholesterol-bearing pullulans (CHP) was determined by contrast variation small-angle neutron scattering. The decomposition of scattering intensities into partial scattering functions of each CHP nanogel component, i.e., pullulan, cholesterol, and the cross-term between the pullulan and the cholesterol allows us to investigate the internal structure of the nanogel. The effective spherical radius of the skeleton formed by pullulan chains was found to be about 8.1 nm. In the CHP nanogel, there are about 19 cross-linking points where a cross-linking point is formed by aggregation of trimer cholesterol molecules with the spatially inhomogeneous distribution of the mass fractal dimension of 2.6. The average radius of the partial chains can also be determined to be 1.7 nm. As the result, the complex structure of the nanogels is coherently revealed at the nanoscopical level.
Noguchi, Yoshifumi*; Hiyama, Miyabi*; Shiga, Motoyuki; Sugino, Osamu*; Akiyama, Hidefumi*
Journal of Physical Chemistry B, 120(34), p.8776 - 8783, 2016/09
Times Cited Count:6 Percentile:13.46(Chemistry, Physical)We investigated the stability of oxyluciferin anions (keto, enol, and enolate isomers) in aqueous solution at room temperature by performing a nanosecond time scale first-principles molecular dynamics simulation. In contrast to all previous quantum chemistry calculations, which suggested the keto-type to be the most stable, we show that the enol-type is slightly more stable than the keto-type, in agreement with some recent experimental studies. The simulation highlights the remarkable hydrophobicity of the keto-type by the cavity formed at the oxyluciferin-water interface as well as a reduction in hydrophobicity with the number of hydrating water molecules. It is therefore predicted that the isomeric form in a hydrated cluster is size-dependent.
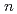 -butyl phosphate in organic and aqueous environments and its relevance to nuclear extraction processes
-butyl phosphate in organic and aqueous environments and its relevance to nuclear extraction processesMu, J.*; Motokawa, Ryuhei; Williams, C. D.*; Akutsu, Kazuhiro*; Nishitsuji, Shotaro*; Masters, A. J.*
Journal of Physical Chemistry B, 120(23), p.5183 - 5193, 2016/06
Times Cited Count:27 Percentile:56.40(Chemistry, Physical)Sekine, Yurina; Fukazawa, Tomoko*; Aizawa, Mamoru*; Kobayashi, Riki*; Chi, S.*; Fernandez-Baca, J. A.*; Yamauchi, Hiroki; Fukazawa, Hiroshi
Journal of Physical Chemistry B, 118(47), p.13453 - 13457, 2014/08
Times Cited Count:4 Percentile:7.91(Chemistry, Physical)Neutron diffraction patterns for deuterated poly-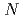 ,,-dimethylacrylamide (PDMAA) hydrogels were measured from 10 to 300 K to investigate the structure and properties of water in the gels. Diffraction peaks observed below 250 K indicate the existence of ice in the hydrogels. Some diffraction peaks from the ice are at lower diffraction angles than those in ordinary hexagonal ice (Ih). These shifts in peaks indicate that the lattice constants of the
,,-dimethylacrylamide (PDMAA) hydrogels were measured from 10 to 300 K to investigate the structure and properties of water in the gels. Diffraction peaks observed below 250 K indicate the existence of ice in the hydrogels. Some diffraction peaks from the ice are at lower diffraction angles than those in ordinary hexagonal ice (Ih). These shifts in peaks indicate that the lattice constants of the 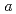 and
and 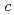 axes in the ice are about 0.29% and 0.3% higher than those in ice Ih, respectively. The results show that bulk low-density ice can exist in PDMAA hydrogels. The distortions in the lattice structure of ice imply significant interactions between water molecules and the surrounding polymer chains, which play an important role in the chemical and mechanical properties of the hydrogel.
axes in the ice are about 0.29% and 0.3% higher than those in ice Ih, respectively. The results show that bulk low-density ice can exist in PDMAA hydrogels. The distortions in the lattice structure of ice imply significant interactions between water molecules and the surrounding polymer chains, which play an important role in the chemical and mechanical properties of the hydrogel.
Yonetani, Yoshiteru; Kono, Hidetoshi
Journal of Physical Chemistry B, 117(25), p.7535 - 7545, 2013/06
Times Cited Count:16 Percentile:37.45(Chemistry, Physical)-butyl phosphate/-octane mixtures by X-ray and neutron scattering in a wide 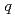 range
rangeMotokawa, Ryuhei; Suzuki, Shinichi; Ogawa, Hiroki*; Antonio, M. R.*; Yaita, Tsuyoshi
Journal of Physical Chemistry B, 116(4), p.1319 - 1327, 2012/02
Times Cited Count:38 Percentile:65.23(Chemistry, Physical)Shimada, Tomoko*; Sakamoto, Naoki*; Motokawa, Ryuhei; Koizumi, Satoshi*; Tirrell, M.*
Journal of Physical Chemistry B, 116(1), p.240 - 243, 2012/01
Times Cited Count:52 Percentile:74.95(Chemistry, Physical)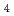 systems (A
systems (A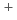 = Li, Na, and K)
= Li, Na, and K)Pauvert, O.*; Salanne, M.*; Zanghi, D.*; Simon, C.*; Reguer, S.*; Thiaudi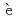 re, D.*; Okamoto, Yoshihiro; Matsuura, Haruaki*; Bessada, C.*
re, D.*; Okamoto, Yoshihiro; Matsuura, Haruaki*; Bessada, C.*
Journal of Physical Chemistry B, 115(29), p.9160 - 9167, 2011/07
Times Cited Count:77 Percentile:84.54(Chemistry, Physical)The structure of molten AF-ZrF system (A=Li, Na, K) is studied using EXAFS spectroscopy with molecular dynamics simulations. From the Zr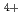 solvation shell point of view, we observe a progressive stabilization of the 7-fold and then of the 6-fold coordinated complexes when passing from Li to Na and K as a "counterion". Particular attention is given to the systems consisting of 35 mol% of ZrF. At that particular composition, the ZrF
solvation shell point of view, we observe a progressive stabilization of the 7-fold and then of the 6-fold coordinated complexes when passing from Li to Na and K as a "counterion". Particular attention is given to the systems consisting of 35 mol% of ZrF. At that particular composition, the ZrF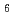
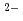 complex predominates largely whatever the nature of the alkali. The calculated vibrational properties of this complex are in excellent agreement with a previous Raman spectroscopy experiment on molten KF-ZrF. The most important differences are observed for the lifetime of these octahedral units. On a larger scale, an intense first sharp diffraction peak is observed for the Zr-Zr partial structure factor, which can be attributed to the correlations between the octahedral units formed.
complex predominates largely whatever the nature of the alkali. The calculated vibrational properties of this complex are in excellent agreement with a previous Raman spectroscopy experiment on molten KF-ZrF. The most important differences are observed for the lifetime of these octahedral units. On a larger scale, an intense first sharp diffraction peak is observed for the Zr-Zr partial structure factor, which can be attributed to the correlations between the octahedral units formed.
Murakami, Hiroshi; Nishi, Takaki*; Toyota, Yuji*
Journal of Physical Chemistry B, 115(19), p.5877 - 5885, 2011/04
Times Cited Count:18 Percentile:39.36(Chemistry, Physical)We present a method based on near-infrared absorption spectroscopy of the OH stretching vibration band of water around 3400 cm to examine if the aqueous cavity size of a protein-unfilled reverse micelle is affected by solubilization of protein, and it has been found for AOT (= bis (2-ethylhexyl) sulfosuccinate) reverse micellar solution with myoglobin that it does not change before and after solubilization of the protein in the water-to-surfactant molar ratio (
to examine if the aqueous cavity size of a protein-unfilled reverse micelle is affected by solubilization of protein, and it has been found for AOT (= bis (2-ethylhexyl) sulfosuccinate) reverse micellar solution with myoglobin that it does not change before and after solubilization of the protein in the water-to-surfactant molar ratio (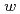
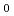 ) from 2 to
) from 2 to  18, that is, the values of the protein-filled and unfilled reverse micelles are the same as that of the reverse micellar solution regardless of size relation between the aqueous cavity of the unfilled reverse micelle and the protein. On the basis of this experimental fact, we propose a model to determine the structural parameters of protein-filled reverse micelles, such as the aqueous cavity radius and the aggregation number of surfactant molecules constituting the micelle, and the molar concentration of the unfilled reverse micelle that exists with the protein-filled reverse micelle in the reverse micellar solution, and derive their values for AOT reverse micellar solution with myoglobin in the range from 2 to 24. On the other hand, circular dichroism measurements and UV-visible absorption spectroscopy of myoglobin/AOT reverse micellar solution and myoglobin/AOT aqueous solution were carried out in order to examine the conformational state of myoglobin in the reverse micellar solution. These experimental results lead to the conclusion that myoglobin is located in the aqueous cavity of the reverse micelle, although the conformational state of the protein is to some extent distorted because of the interaction with AOT compared with that of native myoglobin. Finally, it is suggested that the proposed model is appropriate for reverse micellar solution with a hydrophilic protein molecule that is located in the aqueous cavity of the reverse micelle.
18, that is, the values of the protein-filled and unfilled reverse micelles are the same as that of the reverse micellar solution regardless of size relation between the aqueous cavity of the unfilled reverse micelle and the protein. On the basis of this experimental fact, we propose a model to determine the structural parameters of protein-filled reverse micelles, such as the aqueous cavity radius and the aggregation number of surfactant molecules constituting the micelle, and the molar concentration of the unfilled reverse micelle that exists with the protein-filled reverse micelle in the reverse micellar solution, and derive their values for AOT reverse micellar solution with myoglobin in the range from 2 to 24. On the other hand, circular dichroism measurements and UV-visible absorption spectroscopy of myoglobin/AOT reverse micellar solution and myoglobin/AOT aqueous solution were carried out in order to examine the conformational state of myoglobin in the reverse micellar solution. These experimental results lead to the conclusion that myoglobin is located in the aqueous cavity of the reverse micelle, although the conformational state of the protein is to some extent distorted because of the interaction with AOT compared with that of native myoglobin. Finally, it is suggested that the proposed model is appropriate for reverse micellar solution with a hydrophilic protein molecule that is located in the aqueous cavity of the reverse micelle.
systemPauvert, O.*; Zanghi, D.*; Salanne, M.*; Simon, C.*; Rakhamatullin, A.*; Matsuura, Haruaki*; Okamoto, Yoshihiro; Vivet, F.*; Bessada, C.*
Journal of Physical Chemistry B, 114(19), p.6472 - 6479, 2010/05
Times Cited Count:65 Percentile:80.51(Chemistry, Physical)The structure of the molten LiF-ZrF system up to 50 mol% ZrF was investigated by combining high-temperature nuclear magnetic resonance (NMR) and extended X-ray absorption fine structure (EXAFS) experiments with molecular dynamics (MD) calculations. 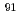 Zr high-temperature NMR experiments give an average coordination of 7 for the zirconium ion on all domains of composition. MD simulations, in agreement with EXAFS experiments at the K-edge of Zr, provide evidence for the coexistence of three different Zr-based complexes, [ZrF], [ZrF
Zr high-temperature NMR experiments give an average coordination of 7 for the zirconium ion on all domains of composition. MD simulations, in agreement with EXAFS experiments at the K-edge of Zr, provide evidence for the coexistence of three different Zr-based complexes, [ZrF], [ZrF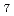 ]
]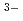 , and [ZrF
, and [ZrF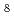 ]
]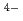 , in the melt.
, in the melt.
Matsuura, Toyoaki*; Ikeda, Hitoe*; Idota, Naokazu*; Motokawa, Ryuhei; Hara, Yoshiaki*; Annaka, Masahiko*
Journal of Physical Chemistry B, 113(51), p.16314 - 16322, 2009/12
Times Cited Count:14 Percentile:32.29(Chemistry, Physical)Fujii, Kentaro; Shikazono, Naoya; Yokoya, Akinari
Journal of Physical Chemistry B, 113(49), p.16007 - 16015, 2009/11
Times Cited Count:19 Percentile:41.91(Chemistry, Physical)In order to verify the possibility of selective damage induction in DNA, the yields of base lesions as well as strand breaks have been measured in dry plasmid DNA films irradiated with highly monochromatized soft X-rays in the energy region of 270-760 eV, which includes the carbon, nitrogen and oxygen K-edges. Our results strongly suggest that (1) the K-shell ionization of oxygen in both the nucleobases as well as in other parts of DNA and in the hydrating water molecules bound to DNA, but not the K-shell ionization of nitrogen in the nucleobases, most likely contributes to the induction of nucleobase lesions, and (2) migration of electrons and holes are involved differentially in the production of each type of DNA lesion. These results could potentially lead to new methods for selective induction of specific types of DNA damage through tuning the energy of soft X-rays.
Jochi, Yasumasa*; Nakagawa, Hiroshi; Kataoka, Mikio; Kitao, Akio*
Journal of Physical Chemistry B, 112(11), p.3522 - 3528, 2008/03
Times Cited Count:11 Percentile:26.89(Chemistry, Physical)Molecular dynamics simulations of crystalline Staphylococcal nuclease in full and minimal hydration states were performed to study hydration effects on protein dynamics at temperatures ranging from 100 to 300 K. In a full hydration state (hydration ratio in weight, h = 0.49), gaps are fully filled with water molecules, whereas only crystal waters are included in a minimal hydration state (h = 0.09). The inflection of the atomic mean-square fluctuation of protein as a function of temperature, known as the glass-like transition, is observed at 220 K in both cases, which is more significant in the full hydration state. By examining the temperature dependence of residual fluctuation, we found that the increase of fluctuations in the loop and terminal regions, which are exposed to water, is much greater than in other regions in the full hydration state, but the mobility of the corresponding regions are relatively restricted in the minimal hydration state by inter-molecular contact. The atomic mean-square fluctuation of water molecules in the full hydration state at 300 K is one order of magnitude greater than that in the minimal hydration state. Above the transition temperature, most water molecules in the full hydration state behave like bulk water, and act as a lubricant for protein dynamics. In contrast, water molecules in the minimal hydration state tend to form more hydrogen bonds with the protein, restricting the fluctuation of these water molecules to the level of the protein. Thus, inter-molecular interaction and solvent mobility are important to understand the glass-like transition in proteins.
Takahashi, Kenji*; Sakai, Shingo*; Tezuka, Hiroaki*; Hiejima, Yusuke*; Katsumura, Yosuke; Watanabe, Masayoshi*
Journal of Physical Chemistry B, 111(18), p.4807 - 4811, 2007/05
Times Cited Count:42 Percentile:68.58(Chemistry, Physical)Kato, Ryuji*; Yoshida, Yoichi*; Katsumura, Yosuke; Takahashi, Kenji*
Journal of Physical Chemistry B, 111(18), p.4770 - 4774, 2007/05
Times Cited Count:54 Percentile:75.95(Chemistry, Physical)