


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Ozawa, Takahiro*; Fukutani, Katsuyuki; 4 of others*
Journal of Physics and Chemistry of Solids, 185, p.111741_1 - 111741_7, 2024/02
Times Cited Count:3 Percentile:34.10(Chemistry, Multidisciplinary)
Nakamura, Jumpei*; Kawakita, Yukinobu; Okabe, Hirotaka*; Li, B.*; Shimomura, Koichiro*; Suemasu, Takashi*
Journal of Physics and Chemistry of Solids, 175, p.111199_1 - 111199_8, 2023/04
Times Cited Count:3 Percentile:14.58(Chemistry, Multidisciplinary)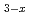 Fe
Fe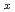 O
O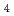 spinel and postspinel with elevating pressure
spinel and postspinel with elevating pressureYamanaka, Takamitsu*; Rahman, S.*; Nakamoto, Yuki*; Hattori, Takanori; Jang, B. G.*; Kim, D. Y.*; Mao, H.-K.*
Journal of Physics and Chemistry of Solids, 167, p.110721_1 - 110721_10, 2022/08
Times Cited Count:1 Percentile:6.72(Chemistry, Multidisciplinary)High-pressure neutron diffraction proved that MnFe O and MnFeO spinels transform into CaMnO-type structure above 18 GPa and 14 GPa, respectively. The transition pressure of MnFeO solutions decreases with increasing Mn content. Synchrotron X-ray M
O and MnFeO spinels transform into CaMnO-type structure above 18 GPa and 14 GPa, respectively. The transition pressure of MnFeO solutions decreases with increasing Mn content. Synchrotron X-ray M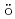 ssbauer experiments revealed that Fe
ssbauer experiments revealed that Fe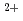 and Fe
and Fe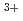 distribution at the tetrahedral (A) and octahedral (B) sites in the spinel structure changes with pressure. MnFeO and MnFeO spinels are ferrimagnetic and the CaMnO-type phase is paramagnetic. The temperature dependence of resistivity indicates that both spinels are semiconductors wherein electrons hop between cations at the A and B sites. A pressure-induced shortening of B-B distance promoted conduction via greater electron mobility between adjacent B cations. The Fe and Fe occupancies at the B sites in MnFeO are much larger than those in MnFeO. The CaMnO-type phase is metallic. Theoretical calculation confirmed the metallic character and Fe d-orbitals strongly renormalized compared to Mn d-orbitals.
distribution at the tetrahedral (A) and octahedral (B) sites in the spinel structure changes with pressure. MnFeO and MnFeO spinels are ferrimagnetic and the CaMnO-type phase is paramagnetic. The temperature dependence of resistivity indicates that both spinels are semiconductors wherein electrons hop between cations at the A and B sites. A pressure-induced shortening of B-B distance promoted conduction via greater electron mobility between adjacent B cations. The Fe and Fe occupancies at the B sites in MnFeO are much larger than those in MnFeO. The CaMnO-type phase is metallic. Theoretical calculation confirmed the metallic character and Fe d-orbitals strongly renormalized compared to Mn d-orbitals.
 Sb
Sb O
O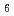 F observed by high-resolution synchrotron and neutron diffraction
F observed by high-resolution synchrotron and neutron diffractionShimono, Seiya*; Ishibashi, Hiroki*; Nagayoshi, Yusuke*; Ikeno, Hidekazu*; Kawaguchi, Shogo*; Hagihara, Masato; Torii, Shuki*; Kamiyama, Takashi*; Ichihashi, Katsuya*; Nishihara, Sadafumi*; et al.
Journal of Physics and Chemistry of Solids, 163, p.110568_1 - 110568_7, 2022/04
Times Cited Count:2 Percentile:14.58(Chemistry, Multidisciplinary)O with electrochemical Li intercalation
intercalationMurase, Satoshi*; Yoshikawa, Yumi*; Fujiwara, Kosuke*; Fukada, Yukimasa*; Teranishi, Takashi*; Kano, Jun*; Fujii, Tatsuo*; Inada, Yasuhiro*; Katayama, Misaki*; Yoshii, Kenji; et al.
Journal of Physics and Chemistry of Solids, 162, p.110468_1 - 110468_6, 2022/03
Times Cited Count:0 Percentile:0.00(Chemistry, Multidisciplinary)We report a trial of the valence control for mixed valence iron triangular oxide YbFeO in order to develop an effective technique to control the frustration of charges in strongly correlated electron systems. The electro-chemical doping of Li into YbFeO was examined on the cell type sample similar to the Li-ion secondary battery cell. Systematic change of the lattice constant, Fe-Fe and Fe-Yb distance were observed with Li doping. Maximum value of the doping was over 300 mAh/g. An EXAFS experiment indicated that Li positioned between Yb octahedron layer (U-layer) and Fe-bipyramidal layer (W-layer). However, detailed change of iron valence state of YbFeO was not clearly observed because of the superimpose of the signal from iron metal nano particles in XANES observation. The results indicate that the electrochemical method might be one of the potential technique to control the frustration of charges in YbFeO.
Hiraka, Haruhiro*; Matsumura, Daiju; Horigane, Kazumasa*; Mizuki, Junichiro*
Journal of Physics and Chemistry of Solids, 150, p.109870_1 - 109870_8, 2021/03
Times Cited Count:0 Percentile:0.00(Chemistry, Multidisciplinary)Miradji, F.; Suzuki, Chikashi; Nakajima, Kunihisa; Osaka, Masahiko
Journal of Physics and Chemistry of Solids, 136, p.109168_1 - 109168_9, 2020/01
Times Cited Count:4 Percentile:20.27(Chemistry, Multidisciplinary)Suzuki, Chikashi; Yaita, Tsuyoshi; Suzuki, Shinichi; Pacold, J.*; Altman, A. B.*; Minasian, S. G.*; Tyliszczak, T.*; Shuk, D. K.*; Yoshida, Hiroyuki; Osaka, Masahiko
Journal of Physics and Chemistry of Solids, 127, p.169 - 177, 2019/04
Times Cited Count:3 Percentile:13.30(Chemistry, Multidisciplinary)A combined method of NEXAFS measurement and DFT-calculation was employed for Cs evaluation in clay minerals. The Cs M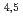 NEXAFS spectra of Cs halides were analyzed using the DFT-calculations, and were well reproduced by incorporating the core-hole strength. The Cs M NEXAFS spectrum of clay minerals was well-reproduced by the DFT-calculations including the major transitions and tail structures with the established method. The further evaluation of this spectrum by charge density suggests that these major transitions and the tail structures likely reflect bonding states and local environments around the Cs atoms. Comparison of electronic states of Cs in the clay mineral with those in the Cs halides by DFT-calculations has shown that the interaction between Cs and the nearest-neighbor atom is largest in the clay mineral, because the energy level of Cs-5s and 5p is closer to that of O-2s and 2p than the s and p orbitals of other alkali metal and alkali earth metal elements.
NEXAFS spectra of Cs halides were analyzed using the DFT-calculations, and were well reproduced by incorporating the core-hole strength. The Cs M NEXAFS spectrum of clay minerals was well-reproduced by the DFT-calculations including the major transitions and tail structures with the established method. The further evaluation of this spectrum by charge density suggests that these major transitions and the tail structures likely reflect bonding states and local environments around the Cs atoms. Comparison of electronic states of Cs in the clay mineral with those in the Cs halides by DFT-calculations has shown that the interaction between Cs and the nearest-neighbor atom is largest in the clay mineral, because the energy level of Cs-5s and 5p is closer to that of O-2s and 2p than the s and p orbitals of other alkali metal and alkali earth metal elements.
Shibata, Hiroki; Hayashi, Hirokazu; Akabori, Mitsuo; Arai, Yasuo; Kurata, Masaki
Journal of Physics and Chemistry of Solids, 75(8), p.972 - 976, 2014/08
Times Cited Count:20 Percentile:60.05(Chemistry, Multidisciplinary)Gibbs free energies of formation of six Ce-Cd intermetallic compounds, CeCd, CeCd, CeCd, CeCd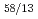 , CeCd and CeCd
, CeCd and CeCd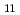 , were evaluated systematically using electrochemical techniques in the temperature range from 673 to 923 K in the LiCl-KCl-CeCl-CdCl molten salt bath. The linear dependence of the Gibbs free energies of formation on temperature yields to the enthalpies and entropies of formation of these intermetallic compounds. By extrapolating the molar Gibbs free energy of Ce-Cd intermetallic compounds to the Cd distillation temperature, it was clear that the molar Gibbs free energy of Ce in Ce-Cd intermetallic compounds decreases gradually from CeCd to CeCd and attains to the minimum value at CeCd. This suggests on the Cd distillation from the U-Pu-Ce-Cd alloy that the dissolution of U or Pu into CeCd should be mostly taken into consideration.
, were evaluated systematically using electrochemical techniques in the temperature range from 673 to 923 K in the LiCl-KCl-CeCl-CdCl molten salt bath. The linear dependence of the Gibbs free energies of formation on temperature yields to the enthalpies and entropies of formation of these intermetallic compounds. By extrapolating the molar Gibbs free energy of Ce-Cd intermetallic compounds to the Cd distillation temperature, it was clear that the molar Gibbs free energy of Ce in Ce-Cd intermetallic compounds decreases gradually from CeCd to CeCd and attains to the minimum value at CeCd. This suggests on the Cd distillation from the U-Pu-Ce-Cd alloy that the dissolution of U or Pu into CeCd should be mostly taken into consideration.
using resonant X-ray emission spectroscopy at the Ba-L and Ti K absorption edgesYoshii, Kenji; Yoneda, Yasuhiro; Jarrige, I.*; Fukuda, Tatsuo; Nishihata, Yasuo; Suzuki, Chikashi; Ito, Yoshiaki*; Terashima, Takahito*; Yoshikado, Shinzo*; Fukushima, Sei*
Journal of Physics and Chemistry of Solids, 75(3), p.339 - 343, 2014/03
Times Cited Count:10 Percentile:39.76(Chemistry, Multidisciplinary)We have studied the electronic properties of ferroelectric BaTiO using two complementary bulk-sensitive spectroscopic probes, resonant X-ray emission spectroscopy (RXES) and X-ray absorption spectroscopy in the partial fluorescence mode (PFY-XAS). Contrary to a previous study, we found no temperature change of a PFY-XAS spectrum at the Ba L edge between room temperature and 150 C. This result is not supportive of the possible presence of the displacement around Ba at the Curie temperature. RXES spectra were measured at the Ti K edge for BaTiO, along with SrTiO and La-doped metallic SrTiO. The photon energy of the emission peak is found to be nearly constant throughout the absorption edge for all three compounds. We deduce the Ti 3d states to have a delocalized character, in contrast with the Ba 5d states, a property which is consistent with the proposed scenario of the formation of electric dipoles in BaTiO.
C. This result is not supportive of the possible presence of the displacement around Ba at the Curie temperature. RXES spectra were measured at the Ti K edge for BaTiO, along with SrTiO and La-doped metallic SrTiO. The photon energy of the emission peak is found to be nearly constant throughout the absorption edge for all three compounds. We deduce the Ti 3d states to have a delocalized character, in contrast with the Ba 5d states, a property which is consistent with the proposed scenario of the formation of electric dipoles in BaTiO.
Suzuki, Chikashi; Nishi, Tsuyoshi; Nakada, Masami; Tsuru, Tomohito; Akabori, Mitsuo; Hirata, Masaru; Kaji, Yoshiyuki
Journal of Physics and Chemistry of Solids, 74(12), p.1769 - 1774, 2013/12
Times Cited Count:14 Percentile:51.43(Chemistry, Multidisciplinary)We investigated the electronic state of a CaF-type (Am,U) mixed oxide using the all-electron full potential linear augmented plane wave method and compared it with those of AmO, AmO, UO, and La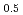 UO. The valence of Am in the mixed oxide was close to that of AmO and the valence of U in the mixed oxide was pentavalent. The electronic structure of AmO was different from that of AmO, particularly just above the Fermi level. In addition, the electronic states of Am and U in the mixed oxide were similar to those of trivalent Am and pentavalent U oxides. These electronic states reflected the high oxygen potential of AmO and the heightened oxygen potential resulting from the addition of Am to UO and also suggested the occurrence of charge transfer from Am to U in the solid solution process.
UO. The valence of Am in the mixed oxide was close to that of AmO and the valence of U in the mixed oxide was pentavalent. The electronic structure of AmO was different from that of AmO, particularly just above the Fermi level. In addition, the electronic states of Am and U in the mixed oxide were similar to those of trivalent Am and pentavalent U oxides. These electronic states reflected the high oxygen potential of AmO and the heightened oxygen potential resulting from the addition of Am to UO and also suggested the occurrence of charge transfer from Am to U in the solid solution process.
Hakoda, Teruyuki; Igarashi, Hidetoshi*; Isozumi, Yukihiro*; Yamamoto, Shunya; Aritani, Hirofumi*; Yoshikawa, Masahito
Journal of Physics and Chemistry of Solids, 74(2), p.200 - 204, 2013/02
Times Cited Count:3 Percentile:16.00(Chemistry, Multidisciplinary)The gasochromic property of dehydrogenation-catalyst loaded tungsten trioxide (M/WO) powders was examined in exposure to gaseous cyclohexane under different kinds and contents of catalysts, catalyst temperatures, and cyclohexane concentrations. The change in the intensity of visible lights reflected from the M/WO powders was in situ obtained using a portable visible-light spectrometer. The catalyst of Pt was a catalyst initiating dehydrogenation and the change of reflected light intensity at lower temperatures in comparison with the catalysts of Pd and Rh. Among 0.1, 0.5, and 1wt% Pt/WO powders, the 0.5wt% Pt/WO powders demonstrated large change of reflected lights. The heating of 0.5wt% Pt/WO powders at temperatures higher than 130C was required to visually detect cyclohexane at a concentration of 1v%, lower than the combustion lower limit (1.3v%).
and BaSO; A Resonant X-ray emission study at the Ba-L edgeYoshii, Kenji; Jarrige, I.; Suzuki, Chikashi; Matsumura, Daiju; Nishihata, Yasuo; Yoneda, Yasuhiro; Fukuda, Tatsuo; Tamura, Kazuhisa; Ito, Yoshiaki*; Mukoyama, Takeshi*; et al.
Journal of Physics and Chemistry of Solids, 73(9), p.1106 - 1110, 2012/09
Times Cited Count:12 Percentile:44.69(Chemistry, Multidisciplinary)We have directly probed the Ba 5d states in the ferroelectric barium titanate BaTiO using resonant X-ray emission spectroscopy (RXES) and X-ray absorption spectroscopy in the partial fluorescence mode (PFY-XAS) at the Ba-L edge. The results are compared with those of the non-ferroelectric BaSO. While the RXES spectra point to a localized character for the Ba 5d states in both compounds, the main peak of the PFY-XAS spectrum, corresponding to the dipolar transitions from 2p to 5d, is found to be significantly broader for BaTiO than for BaSO. On the basis of band structure calculations, this broadening is ascribed to strong hybridization between the unoccupied Ba 5d and O 2p states in the ferroelectric. This suggests that the hybridization between the conduction states of the Ba and O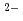 ions, and not only Ti
ions, and not only Ti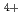 and O, plays a central role in determining the electronic structure of BaTiO.
and O, plays a central role in determining the electronic structure of BaTiO.
Yoshimura, Kimio; Hakoda, Teruyuki; Yamamoto, Shunya; Yoshikawa, Masahito
Journal of Physics and Chemistry of Solids, 73(5), p.696 - 698, 2012/05
Times Cited Count:4 Percentile:19.06(Chemistry, Multidisciplinary)Tungsten trioxide powder with loading 0.1wt% platinum (Pt/WO) was prepared for opticaldetection of organic hydrides such as cyclohexane, decalin by impregnation with PtCl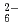 and subsequent calcination in N gas at 500C. The SEM observation of Pt/WO shows that the Pt particles with mean diameters of 80-100 nm were on the surface of the WO powder. The Pt/WO showed coloration for 13% cyclohexane at higher 100C and for 1.3% cyclohexane at 200C. The in-situ XRD results of the Pt/WO in coloring/bleaching change indicate that the coloring of Pt/WO was caused by transformation of WO to tungsten bronze. The analysis of reacted gas demonstrates that Pt on WO produces only hydrogen and benzene through dehydrogenation of cyclohexane over 100C. It was founded that the Pt/WO has potential of optical detection of organic hydrides by heating at higher 100C.
and subsequent calcination in N gas at 500C. The SEM observation of Pt/WO shows that the Pt particles with mean diameters of 80-100 nm were on the surface of the WO powder. The Pt/WO showed coloration for 13% cyclohexane at higher 100C and for 1.3% cyclohexane at 200C. The in-situ XRD results of the Pt/WO in coloring/bleaching change indicate that the coloring of Pt/WO was caused by transformation of WO to tungsten bronze. The analysis of reacted gas demonstrates that Pt on WO produces only hydrogen and benzene through dehydrogenation of cyclohexane over 100C. It was founded that the Pt/WO has potential of optical detection of organic hydrides by heating at higher 100C.
Suzuki, Chikashi; Nishi, Tsuyoshi; Nakada, Masami; Akabori, Mitsuo; Hirata, Masaru; Kaji, Yoshiyuki
Journal of Physics and Chemistry of Solids, 73(2), p.209 - 216, 2012/02
Times Cited Count:28 Percentile:68.65(Chemistry, Multidisciplinary)The authors investigated theoretically core-hole effects on X-ray absorption near-edge structures (XANES) of Np and Am L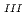 in neptunium dioxide (NpO) and americium dioxide (AmO) with CaF-type crystal lattices using the all-electron full-potential linearized augmented plane-wave (FP-LAPW) method. The peak creation mechanism of XANES was shown by examining the electronic structures of these oxides, which indicated that core-hole screening was more marked for AmO than for NpO because of the difference in the charge transfer between these oxides. Furthermore, the results of charge density analysis suggested that the white line was assigned to the quasi-bound state composed of the localized Np d or Am d components and O components, and that the tail structure was created as a result of delocalized standing waves between the Np or Am atoms.
in neptunium dioxide (NpO) and americium dioxide (AmO) with CaF-type crystal lattices using the all-electron full-potential linearized augmented plane-wave (FP-LAPW) method. The peak creation mechanism of XANES was shown by examining the electronic structures of these oxides, which indicated that core-hole screening was more marked for AmO than for NpO because of the difference in the charge transfer between these oxides. Furthermore, the results of charge density analysis suggested that the white line was assigned to the quasi-bound state composed of the localized Np d or Am d components and O components, and that the tail structure was created as a result of delocalized standing waves between the Np or Am atoms.
Tsutsui, Kenji; Toyama, Takami*; Maekawa, Sadamichi
Journal of Physics and Chemistry of Solids, 72(5), p.354 - 357, 2011/05
Times Cited Count:0 Percentile:0.00(Chemistry, Multidisciplinary)We carry out the numerically exact diagonalization on small clusters of the d-p model with a Ni impurity site to simulate a resonant inelastic X-ray scattering spectra, as well as the single-particle excitation ones. We propose the resonant inelastic X-ray scattering experiment for Ni K edge to confirm the hole binding around the Ni impurity.
adsorbed on Au surfaceSuzuki, Chikashi; Nakagiri, Toshio
Journal of Physics and Chemistry of Solids, 72(1), p.10 - 16, 2011/01
Times Cited Count:5 Percentile:23.95(Chemistry, Multidisciplinary)We evaluated the adsorption of SO molecule on the Au (111) surface using the first-principles calculations by a slab model with a periodic boundary condition. We find that there are six stable adsorption configurations on the Au surface, where SO molecules are adsorbed above the three-fold fcc and hcp hallow sites and atop site. In two of these configurations, S and two O atoms are bound to the Pt atoms, and in two other of them, all the three O atoms are bound to Pt surface atoms, and then, S atom is bound to Au surface atom on the atop site and O atoms are situated on the hallow sites. In addition, it is found that molecular orbitals of SO and those of Au surface are hybridized in the active metal d-bands region, that the localized molecular orbitals in SO are stabilized, and that the charge is transferred from Au to S 3p by SO adsorption on Au surface though the other interaction of S and O (bound to Au) component with Au is little. In addition, the bond between S and O bound to Au become weak by SO adsorption on Au surface because the charge polarization to O-Au bond weakens the bond between S and O bound to Au. This interaction is assumed to encourage the breakage of S-O bond.
Iikubo, Satoshi*; Koyanaka, Hideki*; Shamoto, Shinichi; Takeuchi, Ken*; Kohara, Shinji*; Kodama, Katsuaki; Loong, C.-K.*
Journal of Physics and Chemistry of Solids, 71(11), p.1603 - 1608, 2010/11
Times Cited Count:8 Percentile:36.06(Chemistry, Multidisciplinary)The local crystal structure of dried and deuterated nano-manganese-oxide powder samples was studied via atomic pair distribution function analysis of X-ray and neutron powder diffraction data. The protonated sample shows ultrahigh efficiency as a gold adsorbent even from ppt-level aqueous solutions such as seawater. We show that the nano-manganese-oxide particles have an R-MnO-type local crystal structure. The possible role of the protons on the surface of the nano-particles is discussed.
Fujishita, Hideshi*; Hayashi, Makoto*; Kanai, Takashi*; Yamada, Takahiro*; Igawa, Naoki; Kihara, Kuniaki*
Journal of Physics and Chemistry of Solids, 71(9), p.1285 - 1289, 2010/09
Times Cited Count:3 Percentile:16.24(Chemistry, Multidisciplinary)Atomic displacements are order parameters for displacive structural phase transitions. The temperature evolution of Si atomic displacement in quartz was analyzed by studying the quantum expansion of the Landau potential. The expansion was found to accurately describe the evolution of the atomic displacement over the entire temperature interval. A proportional relationship between spontaneous strain and the square of the atomic displacement was observed over the entire temperature interval.
motion in carbon dioxide deuterohydrate determined by applying maximum entropy method to neutron powder diffraction dataIgawa, Naoki; Taguchi, Tomitsugu; Hoshikawa, Akinori*; Fukazawa, Hiroshi; Yamauchi, Hiroki; Utsumi, Wataru; Ishii, Yoshinobu*
Journal of Physics and Chemistry of Solids, 71(6), p.899 - 905, 2010/06
Times Cited Count:10 Percentile:42.03(Chemistry, Multidisciplinary)Crystal structures of carbon dioxide deuterohydrate were studied by neutron powder diffraction at temperatures from 10 to 200 K. Maps of scattering length density distribution were obtained using a maximum entropy method, which clarified the motion of CO molecules in the hydrate. In small cages, the carbon atom of the CO molecule is at the center of the cage, and the oxygen atoms of CO revolve freely around the carbon atom. In large cages, the carbon atom also is at the center of the cage, but the oxygen atoms tend to revolve around the carbon atom along the plane parallel to the hexagonal facets of the cage.
