


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Xianglian*; Sakuma, Takashi*; Mohapatra, S. R.*; Uehara, Hiroyuki*; Takahashi, Haruyuki*; Kamishima, Osamu*; Igawa, Naoki
Molecular Simulation, 38(5), p.448 - 451, 2012/04
Times Cited Count:4 Percentile:12.31(Chemistry, Physical)Diffuse neutron scattering measurements have been performed on powder PbTe at 10 and 294 K. Oscillatory diffuse scattering intensity is clearly observed at 294 K. Oscillatory form of the diffuse neutron scattering intensity for PbTe was explained on inclusion of correlation effects among thermal displacements of atoms. From the values of correlation effects and Debye-Waller temperature parameter, force constants among the first nearest neighboring atoms was calculated, 
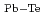 = 215 eV/nm
= 215 eV/nm , and the second ones were
, and the second ones were 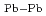 =108 eV/nm and
=108 eV/nm and 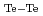 = 144 eV/nm at 294 K.
= 144 eV/nm at 294 K.
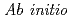 quantum mechanical/molecular mechanical molecular dynamics using multiple-time-scale approach and perturbation theory
quantum mechanical/molecular mechanical molecular dynamics using multiple-time-scale approach and perturbation theoryShiga, Motoyuki; Tachikawa, Masanori*
Molecular Simulation, 33(1-2), p.171 - 184, 2007/02
A new computational method is proposed for quantum-mechanical/molecular-mechanical (QM/MM) molecular dynamics (MD) which is limited to time-independent thermodynamic analysis. The idea is to use the mass-scaling method combined with multiple-time-scale algorithm and an approximate QM/MM Hamiltonian derived from the first-order Rayleigh-Schroedinger perturbation theory in which the electronic polarization is neglected as a first approximation. If the polarization effect is not so strong, the correction can also be considered after the simulation run using the weighted sampling method. The advantage and disadvantage of the method is discussed in terms of its computational efficiency and accuracy. As a simple example, we demonstrate an MD simulation of liquid water containing 1 QM molecule and 255 MM molecules, and discuss the advantages in calculating statistical averages such as radial distribution and heat of solution.
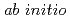 path integral molecular dynamics simulation
path integral molecular dynamics simulationHayashi, Aiko*; Shiga, Motoyuki; Tachikawa, Masanori*
Molecular Simulation, 33(1-2), p.185 - 188, 2007/02
Times Cited Count:3 Percentile:8.85(Chemistry, Physical)Ab initio path integral molecular dynamics simulation has been performed to investigate the lithium-hydrogen bond and dihydrogen bond of C H
H LiH complex. It has been found from the simulation that these unusual types of hydrogen bonds have large geometrical isotope effect which is similar to that of the conventional hydrogen bonding.
LiH complex. It has been found from the simulation that these unusual types of hydrogen bonds have large geometrical isotope effect which is similar to that of the conventional hydrogen bonding.
Yonetani, Yoshiteru*; Kono, Hidetoshi; Fujii, Satoshi*; Sarai, Akinori*; Go, Nobuhiro
Molecular Simulation, 33(1-2), p.103 - 107, 2007/01
Times Cited Count:5 Percentile:16.13(Chemistry, Physical)DNA tetramer sequences AATT and TTAA are known to be conformationally more rigid and flexible, respectively. In this study, we carry out molecular dynamics simulations of these two sequences, and investigate the characteristic hydration pattern. The rigid AATT is found to be more likely to construct the hydration spine in the minor groove than the flexible TTAA. The result suggests that the hydration water molecules play a critical role for determining the sequence dependent deformability of DNA conformation.
; Chihara, Junzo
Molecular Simulation, 16, p.31 - 46, 1996/00
Times Cited Count:5 Percentile:20.84(Chemistry, Physical)no abstracts in English
;
Molecular Simulation, 12(3-6), p.435 - 439, 1994/00
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)no abstracts in English
;
Molecular Simulation, 12(3-6), p.421 - 430, 1994/00
Times Cited Count:3 Percentile:14.68(Chemistry, Physical)no abstracts in English
Chihara, Junzo;
Molecular Simulation, 12(3-6), p.187 - 195, 1994/00
Times Cited Count:3 Percentile:14.68(Chemistry, Physical)no abstracts in English
; Yokokawa, Mitsuo
Molecular Simulation, 12(3-6), p.441 - 444, 1994/00
Times Cited Count:3 Percentile:26.94(Chemistry, Physical)no abstracts in English
