


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Okumura, Masahiko
Journal of Electronic Materials, 54(9), p.7015 - 7026, 2025/09
The machine learning molecular dynamics (MLMD) method enables simulations with high prediction accuracy at low computational cost by learning the results of first-principles calculations (quantum mechanical calculations) using artificial neural networks. This presentation will show how machine-learning molecular dynamics can simulate materials with complex structures. Our MLMD simulations succeeded in reproducing the experimental results of the phonon spectrum of the hydroxy groups of a clay mineral (kaolinite), the superionic transition of thorium dioxide, and the medium-range ordered structure of silica glass, which are difficult to accurately evaluate using other simulation methods, such as classical molecular dynamics.
Kobayashi, Keita; Nakamura, Hiroki; Okumura, Masahiko; Itakura, Mitsuhiro; Machida, Masahiko
Journal of Chemical Physics, 162(24), p.244508_1 - 244508_11, 2025/06
Times Cited Count:0 Percentile:0.00(Chemistry, Physical)The specific heat anomaly in (anti-)fluorite structures was analyzed using machine learning molecular dynamics (MLMD) methods. By employing the Farthest Point Sampling method and the Bootstrap method, first-principles training data were efficiently generated, and machine learning potentials were created for thorium dioxide (fluorite structure) and lithium oxide (anti-fluorite structure). As a result, the MLMD method accurately reproduced the reported thermal properties of thorium dioxide and lithium oxide. These materials exhibit a specific heat anomaly at high temperatures due to sublattice disordering. However, the details are complex and not fully understood. In this study, by applying a local order parameter methodology, which has been used in the analysis of liquid-liquid phase transitions, we revealed that the anomalous specific heat in (anti-)fluorite structures can be interpreted as a consequence of local symmetry breaking.
Shiotani, Kohei; Niiyama, Tomoaki*; Shimokawa, Tomotsugu*
Materials Transactions, 66(6), p.704 - 711, 2025/04
Times Cited Count:0 Percentile:0.00(Materials Science, Multidisciplinary)no abstracts in English
Kobayashi, Keita
New Glass, 39(2), p.13 - 17, 2024/07
The atomic arrangement in glass structures lacks periodicity, and the information obtained experimentally reflects an average structure. Therefore, to estimate the three-dimensional structure of glass materials, molecular dynamics simulations are effective. The results of molecular dynamics calculations strongly depend on the interatomic potential. We have created a machine learning potential (MLP) trained on first-principles calculation results for silica materials. This paper outlines our research of structural analysis of high-density silica glass using machine learning molecular dynamics (MLMD) with the MLP. The MLMD successfully reproduced the experimental data of silica glass. Furthermore, it was revealed that changes in the medium-range order structure in high-density silica glass are characterized by the deformation behavior of ring structures within the Si-O covalent bond network due to the compression.
Sasa, Narimasa
JSIAM Letters, 16, p.37 - 40, 2024/06
Numerical properties of the momentum conservation law for Hamiltonian PDEs are investigated based on a symplectic time integration. In the nonlinear Klein-Gordon system, it is shown that the critical value of the coefficient of the dispersion term is nearly proportional to the inverse square of the total grid number. The result is consistent with the scaling law. On the other hand, in the nonlinear Schr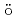 dinger-type system, the critical value of the coefficient does not follow the scaling law.
dinger-type system, the critical value of the coefficient does not follow the scaling law.
 -Fe
-FeSuzudo, Tomoaki; Ebihara, Kenichi; Tsuru, Tomohito; Mori, Hideki*
Journal of Applied Physics, 135(7), p.075102_1 - 075102_7, 2024/02
Times Cited Count:3 Percentile:52.00(Physics, Applied)Fracture of body centred cubic (bcc) metals and alloys below the ductile-to-brittle transition temperature is brittle. This is theoretically explained by the notion that the critical stress intensity factor of a given crack front for brittle fracture is smaller than that for plastic deformation; hence, brittle fracture is chosen over plastic deformation. Although this view is true from a macroscopic point of view, such brittle fracture is always accompanied by small-scale plastic deformation in the vicinity of the crack tip, i.e. crack tip plasticity. This short paper investigates the origin of this plasticity using atomistic modeling with a recently developed machine-learning interatomic potential of -Fe. The computational results identified the precursor of crack tip plasticity, i.e. the group of activated atoms dynamically nucleated by fast crack propagation.
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Urata, Shingo*; Suzuya, Kentaro
Scientific Reports (Internet), 13, p.18721_1 - 18721_12, 2023/11
Times Cited Count:18 Percentile:82.34(Multidisciplinary Sciences)The first sharp peak diffraction peak (FSDP) in the structure factor of amorphous materials is thought to reflect the medium-range order structure in amorphous materials, and the structural origin of the FSDP has been a subject of ongoing debate. In this study, we employed machine learning molecular dynamics (MLMD) with nearly first-principles calculation accuracy to investigate the structural origin of the FSDP in high-density silica glass. First, we successfully reproduced various experimental data of high-density silica glass using MLMD. Furthermore, we revealed that the development (or reduction) of the FSDP in high-density silica glass is characterized by the deformation behavior of ring structures in Si-O covalent bond networks under compression.
Massey, D.*; Williams, C. D.*; Mu, J.*; Masters, A. J.*; Motokawa, Ryuhei; Aoyagi, Noboru; Ueda, Yuki; Antonio, M. R.*
Journal of Physical Chemistry B, 127(9), p.2052 - 2065, 2023/03
Times Cited Count:2 Percentile:10.35(Chemistry, Physical)Kobayashi, Keita; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Materia, 62(3), p.175 - 181, 2023/03
no abstracts in English
Yabuuchi, Kiyohiro*; Suzudo, Tomoaki
Journal of Nuclear Materials, 574, p.154161_1 - 154161_6, 2023/02
Times Cited Count:4 Percentile:47.14(Materials Science, Multidisciplinary)In nuclear materials, irradiation defects cause degradation of mechanical properties. In these materials, the relationship between dislocations and voids is particularly important for mechanical strength. Although only spherical voids have been studied in the past, this study focuses on faceted voids, which are observed simultaneously with spherical voids. In the current study, molecular dynamics was used to analyze the effect of faceted voids in the irradiation hardening of pure iron. Specifically, we clarified the difference in obstacle strength and interaction processes between spherical voids and faceted voids, and that even faceted voids show differences in interaction depending on their crystallographic arrangement with dislocations.
 - and K-montmorillonite
- and K-montmorilloniteKawakita, Ryohei; Saito, Akito*; Sakuma, Hiroshi*; Anraku, Sohtaro; Kikuchi, Ryosuke*; Otake, Tsubasa*; Sato, Tsutomu*
Applied Clay Science, 231, p.106722_1 - 106722_7, 2023/01
Times Cited Count:3 Percentile:32.42(Chemistry, Physical)Tsugawa, Kiyoto*; Hayakawa, Sho*; Okita, Taira*; Aichi, Masaatsu*; Itakura, Mitsuhiro; Suzuki, Katsuyuki*
Computational Materials Science, 215, p.111806_1 - 111806_8, 2022/12
Times Cited Count:6 Percentile:34.34(Materials Science, Multidisciplinary)Yotsuji, Kenji*; Tachi, Yukio; Sakuma, Hiroshi*; Kawamura, Katsuyuki*
Genshiryoku Bakkuendo Kenkyu (CD-ROM), 29(2), p.63 - 81, 2022/12
The understanding of the swelling phenomenon of montmorillonite is essential to predict the physical and chemical behavior of clay-based barriers in radioactive waste disposal systems. This study investigated the key factors controlling crystalline swelling behavior of montmorillonite with different interlayer counter-ions by molecular dynamics (MD) simulations. On the basis of the comparisons between MD simulated and experimental results, the water content in the interlayer in five homoionic (Na , K, Cs, Ca and Sr) montmorillonite was strongly correlated to the hydration number and the preference of an outer- or inner-sphere complex of each counter-ion. The detailed analysis for these results offer insights that the hydration number is controlled by the hydration free energy, the volume and the distribution of each interlayer counter-ion. The systematic MD simulations with virtually variable parameters clarified that the hydration free energy and the charge of interlayer counter- ions compete as influencing factors, and the control the formation rate of an outer-sphere complex of each counter-ion. The empirical relationships between these key factors will allow essential insights into predicting the swelling behavior of montmorillonite with different interlayer counter-ions.
, K, Cs, Ca and Sr) montmorillonite was strongly correlated to the hydration number and the preference of an outer- or inner-sphere complex of each counter-ion. The detailed analysis for these results offer insights that the hydration number is controlled by the hydration free energy, the volume and the distribution of each interlayer counter-ion. The systematic MD simulations with virtually variable parameters clarified that the hydration free energy and the charge of interlayer counter- ions compete as influencing factors, and the control the formation rate of an outer-sphere complex of each counter-ion. The empirical relationships between these key factors will allow essential insights into predicting the swelling behavior of montmorillonite with different interlayer counter-ions.
Suzudo, Tomoaki; Ebihara, Kenichi; Tsuru, Tomohito; Mori, Hideki*
Scientific Reports (Internet), 12, p.19701_1 - 19701_10, 2022/11
Times Cited Count:12 Percentile:58.98(Multidisciplinary Sciences)Body-centered-cubic (bcc) transition metals, such as -Fe and W, cleave along the {100} plane, even though the surface energy is the lowest along the {110} plane. To unravel the mechanism of this odd response, large-scale atomistic simulations of curved cleavage cracks of -Fe were conducted in association with stress intensity factor analyses of straight crack fronts using an interatomic potential created by an artificial neural network technique. The study provides novel findings: Dislocations are emitted from the crack fronts along the {110} cleavage plane, and this phenomenon explains why the {100} plane can be the cleavage plane. However, the simple straight crack-front analyses did not yield the same conclusion. It is suggested that atomistic modeling, at sufficiently large scales to capture the inherent complexities of materials using highly accurate potentials, is necessary to correctly predict the mechanical strength. The method adopted in this study is generally applicable to the cleavage problem of bcc transition metals and alloys.
Kobayashi, Keita; Yamaguchi, Akiko; Okumura, Masahiko
Applied Clay Science, 228, p.106596_1 - 106596_11, 2022/10
Times Cited Count:10 Percentile:73.29(Chemistry, Physical)no abstracts in English
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Cooper, M. W. D.*
Scientific Reports (Internet), 12(1), p.9808_1 - 9808_11, 2022/06
Times Cited Count:19 Percentile:74.34(Multidisciplinary Sciences)no abstracts in English
Sasa, Narimasa
Journal of the Physical Society of Japan, 91(5), p.054001_1 - 054001_8, 2022/05
Times Cited Count:1 Percentile:13.66(Physics, Multidisciplinary) TEM observation and MD simulation of frank partial dislocation climbing in Al-Cu alloy
TEM observation and MD simulation of frank partial dislocation climbing in Al-Cu alloyChen, J.*; Yoshida, Kenta*; Suzudo, Tomoaki; Shimada, Yusuke*; Inoue, Koji*; Konno, Toyohiko*; Nagai, Yasuyoshi*
Materials Transactions, 63(4), p.468 - 474, 2022/04
Times Cited Count:3 Percentile:19.21(Materials Science, Multidisciplinary)In situ electron irradiation using high-resolution transmission electron microscopy (HRTEM) was performed to visualize the Frank loop evolution in aluminium-copper (Al-Cu) alloy with an atomic-scale spatial resolution of 0.12 nm. The  HRTEM observation along the [110] direction of the FCC-Al lattice, Frank partial dislocation bounding an intrinsic stacking fault exhibited an asymmetrical climb along the
HRTEM observation along the [110] direction of the FCC-Al lattice, Frank partial dislocation bounding an intrinsic stacking fault exhibited an asymmetrical climb along the  112
112 direction opposed to those in the reference pure Al under an electron irradiation, with a corresponding displacement-per-atom rate of 0.055-0.120 dpa/s. The asymmetrical climb of the partial dislocation was described as pinning effects due to Cu-Cu bonding in Guinier-Preston zones by a molecular dynamics simulation.
direction opposed to those in the reference pure Al under an electron irradiation, with a corresponding displacement-per-atom rate of 0.055-0.120 dpa/s. The asymmetrical climb of the partial dislocation was described as pinning effects due to Cu-Cu bonding in Guinier-Preston zones by a molecular dynamics simulation.
 3(111) and 5(0-13) grain boundaries of -iron
3(111) and 5(0-13) grain boundaries of -ironEbihara, Kenichi; Suzudo, Tomoaki
Metals, 12(4), p.662_1 - 662_10, 2022/04
Times Cited Count:4 Percentile:25.71(Materials Science, Multidisciplinary)Phosphorus atoms in steels accumulate at grain boundaries via thermal and/or irradiation effects and induce grain boundary embrittlement. Quantitative prediction of phosphorus segregation at grain boundaries under various temperature and irradiation conditions is therefore essential for preventing embrittlement. To develop a model of grain boundary phosphorus segregation in -iron, we studied the migration of a phosphorus atom in two types of symmetrical tilt grain boundaries (3[1-10](111) and 5[100](0-13) grain boundaries) using molecular dynamics simulations with an embedded atom method potential. The results revealed that, in the 3 grain boundary, phosphorus atoms migrate three-dimensionally mainly in the form of interstitial atoms, whereas in the 5 grain boundary, these atoms migrate one-dimensionally mainly via vacancy-atom exchanges. Moreover, de-trapping of phosphorus atoms and vacancies was investigated.
Kobayashi, Keita; Nagai, Yuki; Itakura, Mitsuhiro; Shiga, Motoyuki
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
Times Cited Count:14 Percentile:69.54(Chemistry, Physical)no abstracts in English
