


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
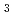 , AgNbO, and KNbO
, AgNbO, and KNbOYoneda, Yasuhiro; Kobayashi, Toru; Tsuji, Takuya; Matsumura, Daiju; Saito, Yuji; Noguchi, Yuji*
Japanese Journal of Applied Physics, 63(9), p.09SP12_1 - 09SP12_10, 2024/09
Times Cited Count:1 Percentile:35.22(Physics, Applied)NaNbO, AgNbO, and KNbO with ABO-type perovskite system is known to possess good ferroelectric properties. We directly determined the rattling space of each atom through local structure analysis. This analysis revealed the bonding sites with large fluctuations varied with varying ion size. Experimental evidence including soft X-ray absorption spectroscopy, indicates that the A-site ions are hybridized with oxygen.
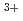 /Eu selectivity with diethylenetriaminepentaacetic acid and its bisamide chelates.
/Eu selectivity with diethylenetriaminepentaacetic acid and its bisamide chelates.Kaneko, Masashi; Sasaki, Yuji; Matsumiya, Masahiko*; Nakase, Masahiko*; Takeshita, Kenji*
Journal of Nuclear Science and Technology, 58(5), p.515 - 526, 2021/05
Times Cited Count:4 Percentile:26.56(Nuclear Science & Technology)Density-functional theory calculations were applied to molecular structure and complex formation reaction modelings of metal ion complexes with diethylenetriaminepentaacetic acid (DTPA) and its bisamide (DTPABA) chelates to understand the metal ions selectivity between Am and Eu. The calculated complexes with DTPA and DTPABA chelates reproduced the coordination geometries of experimental crystal structures. Calculated Gibbs free energies of the complex formation reactions indicated that Am ion forms higher stable complexes with both chelates than Eu ion, being consistent with the experimental results. The higher Am selectivity over Eu was suggested to originate in the larger bond overlap between Am 5f-orbital and N 2s, 2p-orbital. This mean that the covalent contribution between metal ion and donor atoms differentiates the complex formation stabilities, leading to the Am/Eu selectivity. We expect that this study contributes to systematize the origin of metal ions selectivity and to accelerate novel ligands exploration.
Kaneko, Masashi; Suzuki, Hideya; Matsumura, Tatsuro
Inorganic Chemistry, 57(23), p.14513 - 14523, 2018/12
Times Cited Count:24 Percentile:79.50(Chemistry, Inorganic & Nuclear)We elucidated the separation mechanism between Am(III) and Cm(III) ions by using two different types of diamide ligands, diglycolamide (DGA) and alkylated diamide amine (ADAAM), by means of the density functional theory technique and electron density analysis. The molecular geometries and formation reactions of the metal-ligand complexes were modeled by using [M(DGA) ] and [M(ADAAM)(NO)(H
] and [M(ADAAM)(NO)(H O)]. We successfully reproduced Cm(III) selectivity over Am(III) with DGA and Am(III) selectivity over Cm(III) with ADAAM. Furthermore, we analyzed the bonding properties between the metal ion and the diamide-type ligands by using model complexes, [M(DGA)] and [M(ADAAM)(NO)(HO)], and revealed the differences in terms of the bond dissociation energy and the metal 5f-orbital participation in the covalency between the Am(III) and the Cm(III) complexes. It was suggested that the differences were key factors to understand the Am(III)/Cm(III) selectivity.
O)]. We successfully reproduced Cm(III) selectivity over Am(III) with DGA and Am(III) selectivity over Cm(III) with ADAAM. Furthermore, we analyzed the bonding properties between the metal ion and the diamide-type ligands by using model complexes, [M(DGA)] and [M(ADAAM)(NO)(HO)], and revealed the differences in terms of the bond dissociation energy and the metal 5f-orbital participation in the covalency between the Am(III) and the Cm(III) complexes. It was suggested that the differences were key factors to understand the Am(III)/Cm(III) selectivity.
Kimura, Taiki*; Kaneko, Masashi; Watanabe, Masayuki; Miyashita, Sunao*; Nakashima, Satoru*
Dalton Transactions (Internet), 47(42), p.14924 - 14931, 2018/11
Times Cited Count:9 Percentile:43.89(Chemistry, Inorganic & Nuclear)We demonstrated density functional calculations of Eu(III) and Am(III) complexes with pnictogen-donor (X) ligands, CH)X-CH-CH-X(CH) (X = N, P, As and Sb). We investigated the optimized structures of the cmoplexes and the Gibbs energy differences in the complex formation reactions. Those results indicated that the N- and P-donor ligands have Am(III) ion selectivity over Eu(III) ion, especially, the P-donor ligand showed the highest selectivity. The tendency of the Am(III)/Eu(III) selectivity by the pnictogen-dono ligands was found to be comparable to that of soft acid classification in hard and soft acids and bases rule. Mulliken's spin population analysis indicated that the bonding property between the metal ion and the pnictogen atoms correlated with the Am(III)/Eu(III) selectivity. In particular, the participation of f-orbital electrons of the metal ion in the covalency was indicated to have an important role for the selectivity.
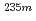 U oxide and fluoride
U oxide and fluorideShigekawa, Yudai*; Kasamatsu, Yoshitaka*; Yasuda, Yuki*; Kaneko, Masashi; Watanabe, Masayuki; Shinohara, Atsushi*
Physical Review C, 98(1), p.014306_1 - 014306_5, 2018/07
Times Cited Count:4 Percentile:32.05(Physics, Nuclear)The nuclear half-life of U has been reported to vary depending on the chemical environment. In this study, both the half-life and the internal-conversion (IC) electron energy spectrum were measured for U with identical chemical environments for the first time. U oxide and fluoride samples were subjected to these measurements, and clear differences in the half-life and the energy spectrum between these samples were observed. The peaks in the energy spectra were identified with the relativistic density functional theory calculation, and the molecular orbital states of the U oxide and fluoride estimated from the energy spectra and the calculation qualitatively explained the difference in the half-lives between the samples.
 ssbauer spectroscopic parameters
ssbauer spectroscopic parametersKaneko, Masashi
Hosha Kagaku, (35), p.36 - 39, 2017/03
This paper is an article for research introduction by winner of the Japan Society of Nuclear and Radiochemical Sciences Encouragement Price 2016. It was mentioned about the achievements which revealed the spin transition behavior of iron complex and the separation mechanism of actinides from lanthanides.
Muramatsu, Yasuji; Hirono, Shigeru*; Umemura, Shigeru*; Ueno, Yuko*; Hayashi, Takayoshi*; Grush, M. M.*; Gullikson, E. M.*; Perera, R. C. C.*
Carbon, 39(9), p.1403 - 1407, 2001/06
Times Cited Count:19 Percentile:58.98(Chemistry, Physical)no abstracts in English
Muramatsu, Yasuji; Ueno, Yuko*; Ishiwata, Yoichi*; Eguchi, Ritsuko*; Watanabe, Masamitsu*; Shin, S.*; Perera, R. C. C.*
Carbon, 39(9), p.1359 - 1402, 2001/06
no abstracts in English
Hirata, Masaru; Bastug, T.*; Tachimori, Shoichi; Sekine, Rika*; Onoe, Jun*; Nakamatsu, Hirohide*
Advances in Quantum Chemistry, Volume 37, p.325 - 333, 2001/00
no abstracts in English
Hirata, Masaru; Tachimori, Shoichi; Sekine, Rika*; Onoe, Jun*; Nakamatsu, Hirohide*
Advances in Quantum Chemistry, Volume 37, p.335 - 351, 2001/00
no abstracts in English
Muramatsu, Yasuji; Takenaka, Hisataka*; Ueno, Yuko*; Gullikson, E. M.*; Perera, R. C. C.*
Applied Physics Letters, 77(17), p.2653 - 2655, 2000/10
Times Cited Count:13 Percentile:50.91(Physics, Applied)no abstracts in English
Kawatsura, Kiyoshi*; Takeshima, Naoki*; Terazawa, Norihisa*; Aoki, Yasushi; Yamamoto, Shunya; Nashiyama, Isamu; Narumi, Kazumasa; Naramoto, Hiroshi
JAERI-Review 99-025, TIARA Annual Report 1998, p.188 - 190, 1999/10
no abstracts in English
Kudo, Hiroshi
Kagaku To Kogyo, 45(5), p.941 - 944, 1992/00
no abstracts in English
Kudo, Hiroshi
Kagaku, 46(11), p.748 - 752, 1991/00
no abstracts in English
Phys.Status Solidi A, 66, p.175 - 181, 1981/00
Times Cited Count:2 Percentile:22.98(Materials Science, Multidisciplinary)no abstracts in English
Kawatsura, Kiyoshi; Ozawa, K.; ;
Phys.Lett.,A, 60(4), p.327 - 329, 1977/04
no abstracts in English
KURRI-TR-55, p.31 - 33, 1969/00
no abstracts in English
Kaneko, Masashi
no journal, ,
Density functional theory (DFT) calculations have been employed to understand the equilibrium structures, electronic states, and stabilities of minor-actinides (MA) and lanthanides (Ln) complexes. An previous application of DFT calculation to the MA/Ln separation has indicated that the Am/Eu selectivity can be explained by the stability in complexation reaction. However, the origin of Am/Eu selectivity remains unclear. This study aims to understand the origin by correlating the stability of their complexes with the covalency in metal-ligand coordination bonds by means of scalar-relativistic DFT calculations. After an optimization of DFT method using benchmark study with Mssbauer spectroscopic parameters, we applied the DFT method to the separation of Am from Eu and analyzed the bonding states between metal and ligands in Eu and Am complexes. As a result, it was found that the wave functions of Eu ion with ligands have syn-phase overlap not depending on donor atoms, whereas those of Am ion have syn-phase overlap in the case of sulfur donors and anti-phase in the case of oxygen donors. This indicates that the difference in bonding states between metal and ligands is an origin in the Am/Eu selectivity.
Yaita, Tsuyoshi
no journal, ,
ssbauer isomer shift and the application to f-block metal coordination chemistryKaneko, Masashi; Watanabe, Masayuki
no journal, ,
Mssbauer isomer shift obtained by measuring Mssbauer spectroscopy has first-order linear responsibility to electron density at nucleus and varies based on the covalent interaction between an element and the surroundings. It assures the quantitative evaluation of the covalent interaction by comparing the linearity. This help us to validate the computational method for estimating the bonding property. We have presented the benchmarking of exchange-correlation functional of density functional theory using Mssbauer isomer shifts of Ru, Os, Eu and Np nuclides. We have indicated that the reproducibility of Mssbauer isomer shift can be correlated to the mixing degree of exact Hartree-Fock exchange admixture in the functional. We will present the applications of the density functional theory to f-block coordination compounds to elucidate their structures and stabilities.
