


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 O
O -pentadentate planar ligands designed for the strongest and selective capture of uranium from seawater
-pentadentate planar ligands designed for the strongest and selective capture of uranium from seawaterMizumachi, Takumi*; Sato, Minami*; Kaneko, Masashi; Takeyama, Tomoyuki*; Tsushima, Satoru*; Takao, Koichiro*
Inorganic Chemistry, 61(16), p.6175 - 6181, 2022/04
Times Cited Count:6 Percentile:52.57(Chemistry, Inorganic & Nuclear)Based on unique 5-fold equatorial coordination of UO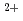 , water-compatible pentadentate planar ligands, Hsaldian and its derivatives, were designed as strong and selective capture of UO in seawater. In the simulated seawater condition (0.5 M NaCl + 2.3 mM HCO
, water-compatible pentadentate planar ligands, Hsaldian and its derivatives, were designed as strong and selective capture of UO in seawater. In the simulated seawater condition (0.5 M NaCl + 2.3 mM HCO /CO
/CO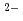 , pH 8), saldian shows the strongest complexation with UO to form UO(saldian) (log
, pH 8), saldian shows the strongest complexation with UO to form UO(saldian) (log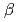
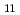 = 28.05
= 28.05 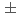 0.07), which is more than 10 order of magnitude greater than amidoxime-based or -inspired ligand systems most commonly employed for U capture from seawater. Good selectivity for UO from other metal ions coexisting in seawater was also demonstrated.
0.07), which is more than 10 order of magnitude greater than amidoxime-based or -inspired ligand systems most commonly employed for U capture from seawater. Good selectivity for UO from other metal ions coexisting in seawater was also demonstrated.
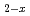 based on experimental data and density functional theory calculation result
based on experimental data and density functional theory calculation resultWatanabe, Masashi; Nakamura, Hiroki; Suzuki, Kiichi; Machida, Masahiko; Kato, Masato
Journal of the American Ceramic Society, 105(3), p.2248 - 2257, 2022/03
Times Cited Count:2 Percentile:9.14(Materials Science, Ceramics)Properties of CeO were evaluated by DFT simulation to determine band gap, Frenkel defect formation energy and defect migration energy. Band gap and Frenkel defect formation energy were used to analyze defect equilibria. Oxygen partial pressure dependence of defect equilibria was evaluated based on oxygen potential experimental data and DFT calculation, and a Brouwer diagram was derived. The defect formation energies, including Frenkel defect, electron-hole pair and so on, were determined and used to evaluate the properties, including oxygen diffusion coefficients, electrical conduction, heat capacity and thermal conductivity. Mechanisms of various properties were discussed for a deeper understanding based on defect chemistry, and the relationship among properties were systematically described.
Kaneko, Masashi
Nihon Genshiryoku Gakkai-Shi ATOMO , 64(1), p.30 - 34, 2022/01
, 64(1), p.30 - 34, 2022/01
Partitioning of minor actinides from rare earths is one of the most important techniques to develop group separation of high-level radioactive liquid waste. In this issue, the results of prediction of separation performance between minor actinides and rare earths observed in solvent extraction and the separation mechanism by means of using density functional theory are explained.
Kaneko, Masashi; Sasaki, Yuji; Wada, Eriko*; Nakase, Masahiko*; Takeshita, Kenji*
Chemistry Letters, 50(10), p.1765 - 1769, 2021/10
Times Cited Count:0 Percentile:0.00(Chemistry, Multidisciplinary)Density functional theory calculation is applied to predict the stability constants for Eu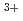 and Am complexes in aqueous solution for molecular modelling of novel separation agents for minor actinides over lanthanides. Logarithm of experimental stability constants correlates with calculated complex formation enthalpies with high reproducibility (R
and Am complexes in aqueous solution for molecular modelling of novel separation agents for minor actinides over lanthanides. Logarithm of experimental stability constants correlates with calculated complex formation enthalpies with high reproducibility (R
 0.98). Prediction of stability constants of novel chelates is demonstrated and indicates a potential availability of the derivatives of diethylenetriaminepentaacetic acid type chelate in acidic condition and enhancement of Am selectivity over Eu.
0.98). Prediction of stability constants of novel chelates is demonstrated and indicates a potential availability of the derivatives of diethylenetriaminepentaacetic acid type chelate in acidic condition and enhancement of Am selectivity over Eu.
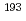 Ir M
Ir M ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adducts
ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adductsKaneko, Masashi; Nakashima, Satoru*
Inorganic Chemistry, 60(17), p.12740 - 12752, 2021/09
Times Cited Count:4 Percentile:29.31(Chemistry, Inorganic & Nuclear)In the present study, density functional theory (DFT) calculation was applied to Vaska's complexes of formula  -[IrCl(CO)(PPh)], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
-[IrCl(CO)(PPh)], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
Fukasawa, Yuto*; Kaneko, Masashi; Nakashima, Satoru*
Journal of Radioanalytical and Nuclear Chemistry, 329(1), p.77 - 84, 2021/07
Times Cited Count:1 Percentile:9.64(Chemistry, Analytical)Density functional theory calculations were applied to understand the selectivity between Am and Eu ions with the crown ethers type ligands. 18C6 is predicted to form the most stable complex with Eu and show the higher stability for Am over Eu, being consistent with previously reported Am/Eu selectivity. We modeled N- and S-donor complexes by using framework of 18C6 complex and analyzed the complexation Gibbs energies, indicating that 18C6 with N-donor atoms is suitable for both complexation and higher Am stability over Eu due to the stronger covalent interaction.
/Eu selectivity with diethylenetriaminepentaacetic acid and its bisamide chelates.Kaneko, Masashi; Sasaki, Yuji; Matsumiya, Masahiko*; Nakase, Masahiko*; Takeshita, Kenji*
Journal of Nuclear Science and Technology, 58(5), p.515 - 526, 2021/05
Times Cited Count:4 Percentile:26.56(Nuclear Science & Technology)Density-functional theory calculations were applied to molecular structure and complex formation reaction modelings of metal ion complexes with diethylenetriaminepentaacetic acid (DTPA) and its bisamide (DTPABA) chelates to understand the metal ions selectivity between Am and Eu. The calculated complexes with DTPA and DTPABA chelates reproduced the coordination geometries of experimental crystal structures. Calculated Gibbs free energies of the complex formation reactions indicated that Am ion forms higher stable complexes with both chelates than Eu ion, being consistent with the experimental results. The higher Am selectivity over Eu was suggested to originate in the larger bond overlap between Am 5f-orbital and N 2s, 2p-orbital. This mean that the covalent contribution between metal ion and donor atoms differentiates the complex formation stabilities, leading to the Am/Eu selectivity. We expect that this study contributes to systematize the origin of metal ions selectivity and to accelerate novel ligands exploration.
complexes of mixed N,O-donor ligands demonstrated on a 10-fold [Eu(TPAMEN)] chelate complexSchnaars, K.; Kaneko, Masashi; Fujisawa, Kiyoshi*
Inorganic Chemistry, 60(4), p.2477 - 2491, 2021/02
Times Cited Count:6 Percentile:43.45(Chemistry, Inorganic & Nuclear)To reduce high-level radiotoxic waste generated by nuclear power plants, highly selective separation agents for minor actinides are mandatory. The mixed N,O-donor ligand 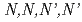 -tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (H
-tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (H TPAEN) has shown good performance as a masking agent in Am/Eu separation studies. In this work, we examine whether a decrease in O-donor basicity can promote the M-N
TPAEN) has shown good performance as a masking agent in Am/Eu separation studies. In this work, we examine whether a decrease in O-donor basicity can promote the M-N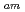 interactions. Therefore, we replace the deprotonated "charged" carboxylic acid groups of TPAEN
interactions. Therefore, we replace the deprotonated "charged" carboxylic acid groups of TPAEN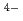 by neutral amide groups and introduce -tetrakis[(6-
by neutral amide groups and introduce -tetrakis[(6-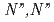 -diethylcarbamoylpyridin-2-yl)methyl]ethylenediamine (TPAMEN) as a new ligand. TPAMEN was crystallized with Eu(OTf) and Eu(NO) 6HO to form positively charged 1:1 [Eu(TPAMEN)] complexes in the solid state. Alterations in the M-O/N bond distances are compared to [Eu(TPAEN)] and investigated by DFT calculations to expose the differences in charge/energy density distributions at europium(III) and the donor functionalities of the TPAEN and TPAMEN. On the basis of estimations of the bond orders, atomic charges spin populations, and density of states in the Eu and potential Am and Cm complexes, the specific contributions of the donor-metal interaction are analyzed. The prediction of complex formation energy differences for the [M(TPAEN)] and [M(TPAMEN)] (M = Eu, Am) complexes provide an outlook on the potential performance of TPAMEN in Am/Eu separation.
-diethylcarbamoylpyridin-2-yl)methyl]ethylenediamine (TPAMEN) as a new ligand. TPAMEN was crystallized with Eu(OTf) and Eu(NO) 6HO to form positively charged 1:1 [Eu(TPAMEN)] complexes in the solid state. Alterations in the M-O/N bond distances are compared to [Eu(TPAEN)] and investigated by DFT calculations to expose the differences in charge/energy density distributions at europium(III) and the donor functionalities of the TPAEN and TPAMEN. On the basis of estimations of the bond orders, atomic charges spin populations, and density of states in the Eu and potential Am and Cm complexes, the specific contributions of the donor-metal interaction are analyzed. The prediction of complex formation energy differences for the [M(TPAEN)] and [M(TPAMEN)] (M = Eu, Am) complexes provide an outlook on the potential performance of TPAMEN in Am/Eu separation.
O)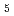 ] with nitrate ions by using density functional theory calculation
] with nitrate ions by using density functional theory calculationKato, Akane*; Kaneko, Masashi; Nakashima, Satoru*
RSC Advances (Internet), 10(41), p.24434 - 24443, 2020/06
Times Cited Count:7 Percentile:27.89(Chemistry, Multidisciplinary)Complexation reactions of ruthenium-nitrosyl complexes in HNO solution were investigated by density functional theory (DFT) calculations in order to predict the stability of Ru species in high-level radioactive liquid waste (HLLW) solution. Equilibrium structure of [Ru(NO)(NO)(HO)] obtained by DFT calculations reproduced the experimental Ru-ligands bond lengths and IR frequencies reported previously. Comparison of the Gibbs energies among the geometrical isomers revealed that the complexation reactions of the ruthenium-nitrosyl complexes with NO proceed via the NO coordination to the equatorial plane toward the Ru-NO axis. We also estimated Gibbs energy differences on the stepwise complexation reactions to succeed in reproducing the fraction of Ru-NO species in 6 M HNO solution, such as in HLLW, by considering the association energy between the Ru-NO species and the substituting ligands. Electron density analyses of the complexes indicated that the strength of the Ru-ligands coordination bonds depends on the stability of the Ru species and the Ru complex without NO at the axial position is more stable than that wit NO, which might attribute to the difference in the trans influence between HO and NO. Finally, we demonstrated the complexation kinetics in the reactions 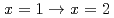 . The present study is expected to enable us to model the precise complexation reactions of platinum-group metals in HNO solution.
. The present study is expected to enable us to model the precise complexation reactions of platinum-group metals in HNO solution.
 Ru Mssbauer parameters of ruthenium-nitrosyl complexes
Ru Mssbauer parameters of ruthenium-nitrosyl complexesKaneko, Masashi; Kato, Akane*; Nakashima, Satoru*; Kitatsuji, Yoshihiro
Inorganic Chemistry, 58(20), p.14024 - 14033, 2019/10
Times Cited Count:12 Percentile:56.06(Chemistry, Inorganic & Nuclear)We applied density functional theory calculations to ruthenium-nitrosyl complexes, which are known to exist in high-level radioactive waste, to give a theoretical correlation between Ru Mssbauer spectroscopic parameters (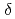 and
and 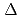
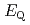 ) and ligand field strength (
) and ligand field strength (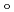 ) for the first time. The structures of the series of complexes, [Ru(NO)L] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO
) for the first time. The structures of the series of complexes, [Ru(NO)L] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO )L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C
)L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C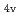 symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of
symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of 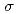 -donor and
-donor and 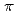 -acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
-acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
Nakashima, Satoru*; Kaneko, Masashi; Yoshinami, Keisuke*; Iwai, Saki*; Dote, Haruka*
Hyperfine Interactions, 239(1), p.39_1 - 39_15, 2018/12
Times Cited Count:2 Percentile:62.30(Physics, Atomic, Molecular & Chemical)The present study reveals the on/off of spin-crossover (SCO) phenomenon in assembled Fe(II) complexes bridged by bis(pyridyl) type ligand. Whether SCO phenomenon occurs or not in assembled Fe(II) complexes bridged by bis(pyridyl) type ligand is determined by local structure around iron atom. SCO phenomenon occurs when the coordinating pyridines facing to each other across the iron atom are propeller type, while the phenomenon does not occur when they are parallel type or distorted propeller type. DFT calculation explained that, in the shortening of Fe-pyridine bonds when changing from high-spin state to low-spin state, the pyridines of propeller type can approach the iron atom with smaller steric hindrance than those of parallel and distorted propeller type complexes. The local structure is controlled by introducing methyl substituent and introducing -system, changing SCO phenomenon. And the transition temperature of SCO is also controlled in assembled complexes bridged by 1,2-bis(4-pyridyl)ethane by mixing anionic ligand.
ssbauer isomer shifts using second-order Douglas-Kroll-Hess HamiltonianKaneko, Masashi; Watanabe, Masayuki; Miyashita, Sunao*; Nakashima, Satoru*
Hyperfine Interactions, 239(1), p.20_1 - 20_10, 2018/12
Times Cited Count:4 Percentile:81.15(Physics, Atomic, Molecular & Chemical)We optimized a mixing ratio of exchange energy between pure DFT and exact Hartree-Fock using TPSS exchange-correlation functional to estimate the accurate coordination bonds in f-block complexes by numerically benchmarking with the experimental data of Mssbauer isomer shifts for 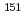 Eu and
Eu and 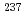 Np nuclides. Second-order Douglas-Kroll-Hess Hamiltonian with segmented all-electron relativistically contracted basis set was employed to calculate the electron densities at Eu and Np nuclei, i.e. contact densities, for each five complexes for Eu(III) and Np(IV) systems. We compared the root mean square deviation values of their isomer shifts between experiment and calculation by changing the mixing ratio of Hartree-Fock exchange parameter from 0 to 100 % at intervals of 10 %. As the result, it was indicated that the mixing ratio of 30 and 60 % for Eu and Np benchmark systems, respectively, gives the smallest deviation values. Mulliken's spin population analysis indicated that the covalency in the metal-ligand bonds for both Eu and Np complexes decreases with increasing the Hartree-Fock exchange admixture.
Np nuclides. Second-order Douglas-Kroll-Hess Hamiltonian with segmented all-electron relativistically contracted basis set was employed to calculate the electron densities at Eu and Np nuclei, i.e. contact densities, for each five complexes for Eu(III) and Np(IV) systems. We compared the root mean square deviation values of their isomer shifts between experiment and calculation by changing the mixing ratio of Hartree-Fock exchange parameter from 0 to 100 % at intervals of 10 %. As the result, it was indicated that the mixing ratio of 30 and 60 % for Eu and Np benchmark systems, respectively, gives the smallest deviation values. Mulliken's spin population analysis indicated that the covalency in the metal-ligand bonds for both Eu and Np complexes decreases with increasing the Hartree-Fock exchange admixture.
Kaneko, Masashi; Suzuki, Hideya; Matsumura, Tatsuro
Inorganic Chemistry, 57(23), p.14513 - 14523, 2018/12
Times Cited Count:24 Percentile:79.50(Chemistry, Inorganic & Nuclear)We elucidated the separation mechanism between Am(III) and Cm(III) ions by using two different types of diamide ligands, diglycolamide (DGA) and alkylated diamide amine (ADAAM), by means of the density functional theory technique and electron density analysis. The molecular geometries and formation reactions of the metal-ligand complexes were modeled by using [M(DGA)] and [M(ADAAM)(NO)(HO)]. We successfully reproduced Cm(III) selectivity over Am(III) with DGA and Am(III) selectivity over Cm(III) with ADAAM. Furthermore, we analyzed the bonding properties between the metal ion and the diamide-type ligands by using model complexes, [M(DGA)] and [M(ADAAM)(NO)(HO)], and revealed the differences in terms of the bond dissociation energy and the metal 5f-orbital participation in the covalency between the Am(III) and the Cm(III) complexes. It was suggested that the differences were key factors to understand the Am(III)/Cm(III) selectivity.
Kimura, Taiki*; Kaneko, Masashi; Watanabe, Masayuki; Miyashita, Sunao*; Nakashima, Satoru*
Dalton Transactions (Internet), 47(42), p.14924 - 14931, 2018/11
Times Cited Count:9 Percentile:43.89(Chemistry, Inorganic & Nuclear)We demonstrated density functional calculations of Eu(III) and Am(III) complexes with pnictogen-donor (X) ligands, CH)X-CH-CH-X(CH) (X = N, P, As and Sb). We investigated the optimized structures of the cmoplexes and the Gibbs energy differences in the complex formation reactions. Those results indicated that the N- and P-donor ligands have Am(III) ion selectivity over Eu(III) ion, especially, the P-donor ligand showed the highest selectivity. The tendency of the Am(III)/Eu(III) selectivity by the pnictogen-dono ligands was found to be comparable to that of soft acid classification in hard and soft acids and bases rule. Mulliken's spin population analysis indicated that the bonding property between the metal ion and the pnictogen atoms correlated with the Am(III)/Eu(III) selectivity. In particular, the participation of f-orbital electrons of the metal ion in the covalency was indicated to have an important role for the selectivity.
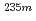 U oxide and fluoride
U oxide and fluorideShigekawa, Yudai*; Kasamatsu, Yoshitaka*; Yasuda, Yuki*; Kaneko, Masashi; Watanabe, Masayuki; Shinohara, Atsushi*
Physical Review C, 98(1), p.014306_1 - 014306_5, 2018/07
Times Cited Count:4 Percentile:32.05(Physics, Nuclear)The nuclear half-life of U has been reported to vary depending on the chemical environment. In this study, both the half-life and the internal-conversion (IC) electron energy spectrum were measured for U with identical chemical environments for the first time. U oxide and fluoride samples were subjected to these measurements, and clear differences in the half-life and the energy spectrum between these samples were observed. The peaks in the energy spectra were identified with the relativistic density functional theory calculation, and the molecular orbital states of the U oxide and fluoride estimated from the energy spectra and the calculation qualitatively explained the difference in the half-lives between the samples.
Kaneko, Masashi; Watanabe, Masayuki
Journal of Radioanalytical and Nuclear Chemistry, 316(3), p.1129 - 1137, 2018/06
Times Cited Count:18 Percentile:83.16(Chemistry, Analytical)We applied density functional theory calculations to Am(III) and Eu(III) complexes with chalcogen-donor ligands of the formula N(EPMe) (E = O, S, Se, Te). We calculated the equilibrium structures and relative stabilities of the complexes in the complexation reaction. The results indicated that the tendency of the relative stability is O 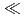 S
S  Se Te, which is consistent with the trend of soft acid classification. Molecular orbital overlap population analysis suggested that this tendency can be correlated with the bonding type in the covalent interaction between the f-orbitals of the metal atom and the chalcogen-donor atoms.
Se Te, which is consistent with the trend of soft acid classification. Molecular orbital overlap population analysis suggested that this tendency can be correlated with the bonding type in the covalent interaction between the f-orbitals of the metal atom and the chalcogen-donor atoms.
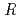 / values by benchmark study of the Mssbauer Isomer shifts for Ru, Os complexes using relativistic DFT calculations
/ values by benchmark study of the Mssbauer Isomer shifts for Ru, Os complexes using relativistic DFT calculationsKaneko, Masashi; Yasuhara, Hiroki*; Miyashita, Sunao*; Nakashima, Satoru*
Hyperfine Interactions, 238(1), p.36_1 - 36_9, 2017/11
Times Cited Count:2 Percentile:75.82(Physics, Atomic, Molecular & Chemical)We aim to evaluate the validity of density functional calculations to the bonding property for Ru and Os complexes. We performed the benchmarking of theoretical computational method with Ru, 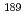 Os Mssbauer isomer shifts. As the result, the computational values of the electron densities at nucleus position correlated with the experimental Mssbauer isomer shifts.
Os Mssbauer isomer shifts. As the result, the computational values of the electron densities at nucleus position correlated with the experimental Mssbauer isomer shifts.
ssbauer isomer shifts with density functional calculationsKaneko, Masashi; Watanabe, Masayuki; Miyashita, Sunao*; Nakashima, Satoru*
Radioisotopes, 66(8), p.289 - 300, 2017/08
Scalar-relativistic density functional calculations applied to some trivalent europium complexes. Five Eu(III) complexes whose Eu Mssbauer isomer shifts vary from -1.8 to 0.5 mm/s are referred by previously reported results. Geometrical optimizations of their complexes reproduces the experimental coordination structures. Single-point calculations are applied to their optimized geometries at three density functionals, namely, BP86, B3LYP, and B2PLYP, to obtain their electron densities at Eu nucleus. A comparison of the linearity between the electron densities and the corresponding Eu Mssbauer isomer shifts reveals that B2PLYP functional shows the best linearity. Electron population and bond analyses indicate that d- and f-orbital electrons of Eu ion in the complexes are found to be correlated to the experimental Eu Mssbauer isomer shifts. This indicates that the d- and f-orbital electrons are involved in the covalent interaction of the coordination bond between the Eu ion and the ligands.
ssbauer spectroscopic parametersKaneko, Masashi
Hosha Kagaku, (35), p.36 - 39, 2017/03
This paper is an article for research introduction by winner of the Japan Society of Nuclear and Radiochemical Sciences Encouragement Price 2016. It was mentioned about the achievements which revealed the spin transition behavior of iron complex and the separation mechanism of actinides from lanthanides.
Kaneko, Masashi; Watanabe, Masayuki; Miyashita, Sunao*; Nakashima, Satoru*
Journal of Nuclear and Radiochemical Sciences (Internet), 17, p.9 - 15, 2017/03
Density functional calculations were applied to the complexation of Eu(III) and Am(III) ions with phosphinic acid (O-donor) and dihiophosphinic acid (S-donor) from the viewpoint of the bonding nature of valence orbitals in metal ion. Two and four conformers for S-donor and O-donor complexes, respectively were optimized. Their stabilization energies by complex formation toward [M(HO)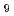 ] were estimated. As the result, the energies reproduced the experimental Am(III)/Eu(III) selectivity that O-donor ligand preferably coordinates to Eu(III) ion, whereas S-donor ligand selectively coordinates to Am(III) ion. Focused on the bonding natures of d and f-orbital electrons, it was indicated that the d-orbital electrons in both Eu and Am complexes participate in the covalency as bonding-type nature and have the almost same contribution. Meanwhile, the contribution of the f-orbital electrons was different between Eu and Am complexes and indicated that in the case of S-donor complex, non-bonding type and bonding type contributions were observed for Eu and Am complexes, respectively and in the case of O-donor complex, bonding type and anti-bonding type contributions were observed for Eu and Am complexes, respectively. This result suggested that the bonding natures of d-orbital electrons contribute to the geometrical similarity of molecular structures for Eu and Am complexes and the bonding natures of f-orbital electrons contribute to the difference in the selectivity of Eu and Am ions.
] were estimated. As the result, the energies reproduced the experimental Am(III)/Eu(III) selectivity that O-donor ligand preferably coordinates to Eu(III) ion, whereas S-donor ligand selectively coordinates to Am(III) ion. Focused on the bonding natures of d and f-orbital electrons, it was indicated that the d-orbital electrons in both Eu and Am complexes participate in the covalency as bonding-type nature and have the almost same contribution. Meanwhile, the contribution of the f-orbital electrons was different between Eu and Am complexes and indicated that in the case of S-donor complex, non-bonding type and bonding type contributions were observed for Eu and Am complexes, respectively and in the case of O-donor complex, bonding type and anti-bonding type contributions were observed for Eu and Am complexes, respectively. This result suggested that the bonding natures of d-orbital electrons contribute to the geometrical similarity of molecular structures for Eu and Am complexes and the bonding natures of f-orbital electrons contribute to the difference in the selectivity of Eu and Am ions.

