


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
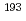 Ir M
Ir M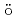 ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adducts
ssbauer spectroscopic parameters of Vaska's complexes and their oxidative adductsKaneko, Masashi; Nakashima, Satoru*
Inorganic Chemistry, 60(17), p.12740 - 12752, 2021/09
Times Cited Count:3 Percentile:30.69(Chemistry, Inorganic & Nuclear)In the present study, density functional theory (DFT) calculation was applied to Vaska's complexes of formula 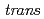 -[IrCl(CO)(PPh
-[IrCl(CO)(PPh )
) ], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
], and their oxidative adducts with small molecules (YZ) including H, i.e., -[IrCl(YZ)(CO))(PPh)], to successfully correlate the electronic states of the complexes with the corresponding Ir Mssbauer spectroscopic parameters. After confirming the reproducibility of the DFT methods for elucidating the equilibrium structures and Ir Mssbauer isomer shifts of the octahedral Ir complexes, the isomer shifts and quadrupole splitting values of Vaska's complexes and their oxidative adducts were calculated. A bond critical point analysis revealed that the tendency in the isomer shifts was correlated with the strength of the covalent interaction in the coordination bonds. In an electric field gradient (EFG) analysis of the oxidative adducts, the sign of the principal axis was found to be positive for the complex with YZ = Cl and negative for the complex with YZ = H. This reversal of the sign of the EFG principal axis was caused by the difference in the electron density distribution for the coordination bonds between Ir and YZ, according to a density of states analysis.
O) ]
]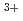 with nitrate ions by using density functional theory calculation
with nitrate ions by using density functional theory calculationKato, Akane*; Kaneko, Masashi; Nakashima, Satoru*
RSC Advances (Internet), 10(41), p.24434 - 24443, 2020/06
Times Cited Count:6 Percentile:30.44(Chemistry, Multidisciplinary)Complexation reactions of ruthenium-nitrosyl complexes in HNO solution were investigated by density functional theory (DFT) calculations in order to predict the stability of Ru species in high-level radioactive liquid waste (HLLW) solution. Equilibrium structure of [Ru(NO)(NO)(HO)] obtained by DFT calculations reproduced the experimental Ru-ligands bond lengths and IR frequencies reported previously. Comparison of the Gibbs energies among the geometrical isomers revealed that the complexation reactions of the ruthenium-nitrosyl complexes with NO proceed via the NO coordination to the equatorial plane toward the Ru-NO axis. We also estimated Gibbs energy differences on the stepwise complexation reactions to succeed in reproducing the fraction of Ru-NO species in 6 M HNO solution, such as in HLLW, by considering the association energy between the Ru-NO species and the substituting ligands. Electron density analyses of the complexes indicated that the strength of the Ru-ligands coordination bonds depends on the stability of the Ru species and the Ru complex without NO at the axial position is more stable than that wit NO, which might attribute to the difference in the trans influence between HO and NO. Finally, we demonstrated the complexation kinetics in the reactions
proceed via the NO coordination to the equatorial plane toward the Ru-NO axis. We also estimated Gibbs energy differences on the stepwise complexation reactions to succeed in reproducing the fraction of Ru-NO species in 6 M HNO solution, such as in HLLW, by considering the association energy between the Ru-NO species and the substituting ligands. Electron density analyses of the complexes indicated that the strength of the Ru-ligands coordination bonds depends on the stability of the Ru species and the Ru complex without NO at the axial position is more stable than that wit NO, which might attribute to the difference in the trans influence between HO and NO. Finally, we demonstrated the complexation kinetics in the reactions 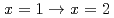 . The present study is expected to enable us to model the precise complexation reactions of platinum-group metals in HNO solution.
. The present study is expected to enable us to model the precise complexation reactions of platinum-group metals in HNO solution.
 Ru Mssbauer parameters of ruthenium-nitrosyl complexes
Ru Mssbauer parameters of ruthenium-nitrosyl complexesKaneko, Masashi; Kato, Akane*; Nakashima, Satoru*; Kitatsuji, Yoshihiro
Inorganic Chemistry, 58(20), p.14024 - 14033, 2019/10
Times Cited Count:12 Percentile:62.43(Chemistry, Inorganic & Nuclear)We applied density functional theory calculations to ruthenium-nitrosyl complexes, which are known to exist in high-level radioactive waste, to give a theoretical correlation between Ru Mssbauer spectroscopic parameters (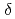 and
and 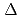
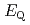 ) and ligand field strength (
) and ligand field strength (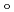 ) for the first time. The structures of the series of complexes, [Ru(NO)L] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO
) for the first time. The structures of the series of complexes, [Ru(NO)L] (L = Br, Cl, NH, CN), were modeled based on the corresponding single-crystal X-ray coordinates. The comparisons of the geometries and total energies between the different spin states suggested that the singlet spin state of [Ru(II)(NO )L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C
)L] complexes were the most stable. The calculated results of both the and values reproduced the experimental results by reported previously and increased in the order of L = Br, Cl, NH, CN. Finally, we estimated the ligand field strength () based on molecular orbitals, assuming C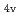 symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of
symmetry and showed the increase of values in that order, being consistent with well-known spectrochemical series of ligands. The increase attributes to the strengthening of the abilities of 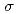 -donor and
-donor and 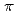 -acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
-acceptor of the L-ligands to the Ru atom, resulting in the increase of the values.
Nakashima, Satoru*; Kaneko, Masashi; Yoshinami, Keisuke*; Iwai, Saki*; Dote, Haruka*
Hyperfine Interactions, 239(1), p.39_1 - 39_15, 2018/12
Times Cited Count:2 Percentile:65.58(Physics, Atomic, Molecular & Chemical)The present study reveals the on/off of spin-crossover (SCO) phenomenon in assembled Fe(II) complexes bridged by bis(pyridyl) type ligand. Whether SCO phenomenon occurs or not in assembled Fe(II) complexes bridged by bis(pyridyl) type ligand is determined by local structure around iron atom. SCO phenomenon occurs when the coordinating pyridines facing to each other across the iron atom are propeller type, while the phenomenon does not occur when they are parallel type or distorted propeller type. DFT calculation explained that, in the shortening of Fe-pyridine bonds when changing from high-spin state to low-spin state, the pyridines of propeller type can approach the iron atom with smaller steric hindrance than those of parallel and distorted propeller type complexes. The local structure is controlled by introducing methyl substituent and introducing -system, changing SCO phenomenon. And the transition temperature of SCO is also controlled in assembled complexes bridged by 1,2-bis(4-pyridyl)ethane by mixing anionic ligand.
Nakamoto, Tadahiro*; Nakada, Masami; Nakamura, Akio
Journal of Nuclear Science and Technology, 39(Suppl.3), p.102 - 105, 2002/11
no abstracts in English
Ru Mssbauer isomer shiftKaneko, Masashi; Kato, Akane*; Nakashima, Satoru*; Kitatsuji, Yoshihiro
no journal, ,
A linear relationship between Mssbauer isomer shift values and electron densities at nucleus position assures the quantitative evaluation of the covalent interaction between a Mssbauer element and its surroundings. The use of this linear relationship makes us to predict a covalency in unknown compounds by electron density analysis based on quantum chemical calculations. As the first step to elucidate stability of ruthenium, which is known to be one of obstructive factors for separation process of high-level radioactive waste, in this study, we performed a fundamental investigation to predict the chemical bonding properties of nitrosylruthenium complexes. We modeled several nitrosylruthenium complexes with basic ligands, such chloride ion and ammonia, namely, [Ru(NO)L] (L = Br, Cl, NH, CN), by density functional calculation and estimated Ru Mssbauer isomer shifts by electron density analysis. The calculated results reproduced the experimental metal-ligand bond lengths and Mssbauer isomer shifts within the error of 0.1 mm/s. We also calculated the orbital splitting values of Ru d-electron. The value increased in the order of L = Br, Cl, NH, CN and correlated to that of Mssbauer isomer shift values. This indicated that the covalent interation between metal and ligand originates in the d-electron contribution of ruthenium to the coordination bonds.
Kaneko, Masashi; Kato, Akane*; Nakashima, Satoru*; Kitatsuji, Yoshihiro; Watanabe, Masayuki
no journal, ,
Ruthenium exists as ruthenium nitrosyls, [Ru(NO)], in high-level radioactive liquid waste and shows various stabilities depending on concentrations of nitrate and hydroxide ions. The detailed stabilities, however, remain to be unclear. As the first step to understand the stabilities of ruthenium nitrosyls, the present study focuses on the structural and bonding properties of the nitrosylruthenium species with fundamental ligands, such as chloride ion and ammonia. We modeled the ruthenium species by referring the corresponding single crystal structures and calculated the stable geometries under aqueous condition. The result reproduced the ruthenium-ligand bond lengths and the stretching vibrational energies of nitrosyl group. We also estimated Ru Mssbauer isomer shifts based on electron density analysis and succeeded in reproducing the ismoer shifts. In the presentation, we will indicate the correlation between the isomer shifts and ligand field splitting derived by molecular orbital analysis and discuss an origin of the stability of ruthenium nitrosyls.
