


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
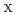 (X = 1, 2)
(X = 1, 2)Kido, Kentaro
International Journal of Quantum Chemistry, 121(21), p.e26781_1 - e26781_15, 2021/11
Times Cited Count:2 Percentile:11.68(Chemistry, Physical)Miyahara, Naoya; Miwa, Shuhei; Horiguchi, Naoki; Sato, Isamu*; Osaka, Masahiko
Journal of Nuclear Science and Technology, 56(2), p.228 - 240, 2019/02
Times Cited Count:9 Percentile:63.15(Nuclear Science & Technology)In order to improve LWR source term under severe accident conditions, the first version of a fission product (FP) chemistry database named "ECUME" was developed. The ECUME is intended to include major chemical reactions and their effective kinetic constants for representative SA sequences. It is expected that the ECUME can serve as a fundamental basis from which FP chemical models in the SA analysis codes can be elaborated. The implemented chemical reactions in the first version were those for representative gas species in Cs-I-B-Mo-O-H system. The chemical reaction kinetic constants were evaluated from either literature data or calculated values using ab-initio calculations. The sample chemical reaction calculation using the presently constructed dataset showed meaningful kinetics effects at 1000 K. Comparison of the chemical equilibrium compositions by using the dataset with those by chemical equilibrium calculations has shown rather good consistency for the representative Cs-I-B-Mo-O-H species. From these results, it was concluded that the present dataset should be useful to evaluate FP chemistry in Cs-I-B-Mo-O-H system under LWA SA conditions.
 potential energy surfaces for the lowest three doublet states (1
potential energy surfaces for the lowest three doublet states (1 A', 2A', and 1A") of the BrH
A', 2A', and 1A") of the BrH system
systemKurosaki, Yuzuru; Takayanagi, Toshiyuki
Journal of Chemical Physics, 119(15), p.7838 - 7856, 2003/10
Times Cited Count:26 Percentile:62.87(Chemistry, Physical)Adiabatic potential energy surfaces of the lowest three doublet states (1A', 2A', and 1A") for the BrH system have been calculated globally using the MRCI+Q method with the aug-cc-pVTZ basis set. Spin-orbit effects were considered on the basis of Breit-Pauli Hamiltonian. The calculated adiabatic energies were fitted to the analytical functional form of many-body expansion. The barrier heights of the abstraction and exchange reactions on the ground-state PES were calculated to be 1.28 and 11.71 kcal mol , respectively, at the MRCI+Q/aug-cc-pVTZ level of theory. The fits for the three PESs were successful within the accuracy of 0.1 kcal mol. Thermal rate constants for the abstraction and exchange reactions and their isotopic variants were calculated with the fitted 1A' PES using the ICVT/LAG method. The calculated rate constants for the abstarction reactions agree fairly well with experiment but those for the exchange reactions were much smaller than experiment, which suggests that the reliable experimental data are still insufficient.
, respectively, at the MRCI+Q/aug-cc-pVTZ level of theory. The fits for the three PESs were successful within the accuracy of 0.1 kcal mol. Thermal rate constants for the abstraction and exchange reactions and their isotopic variants were calculated with the fitted 1A' PES using the ICVT/LAG method. The calculated rate constants for the abstarction reactions agree fairly well with experiment but those for the exchange reactions were much smaller than experiment, which suggests that the reliable experimental data are still insufficient.
H + Cl
+ Cl  CHCl + Cl reaction; Ab initio molecular orbital study
CHCl + Cl reaction; Ab initio molecular orbital studyKurosaki, Yuzuru
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
The CASSCF and MRCI calculations with the cc-pVTZ basis set have been carried out for the CH + Cl CHCl + Cl reaction. It has been revealed that the reaction has a small barrier from the CHCl + Cl side at the CASSCF level of theory, but it has no barrier at the MRCI level. Namely, the CHCl + Cl CH + Cl reaction was predicted to be a spontaneous reaction. The result of the MRCI calculation strongly supports the prediction of our previous PMP4(SDTQ) calculation [J. Mol. Struct. (Theochem) 503 (2000) 231].
Ikezoe, Yasumasa; Suzuki, Kazuya; Nakashima, Mikio; Yokoyama, Atsushi; Shiraishi, Hirotsugu; Ono, Shinichi*
JAERI-Research 98-051, 43 Pages, 1998/09
 JAERI-Research-98-051.pdf:1.63MB
JAERI-Research-98-051.pdf:1.63MB
no abstracts in English
; Yokoyama, Keiichi; Noda, Kenji
Journal of Nuclear Materials, 258-263, p.571 - 575, 1998/00
Times Cited Count:14 Percentile:72.44(Materials Science, Multidisciplinary)no abstracts in English
CN molecule; An Experimental and ab initio studyKudo, Hiroshi; Hashimoto, Masashi; Yokoyama, Keiichi; Wu, C. H.*; A.E.Dorigo*; F.M.Bickelhaupt*; P.von-R.Schleyer*
Journal of Chemical Physics, 99(17), p.6477 - 6482, 1995/00
no abstracts in English
