


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Wu, P.*; Murai, Naoki; Li, T.*; Kajimoto, Ryoichi; Nakamura, Mitsutaka; Kofu, Maiko; Nakajima, Kenji; Xia, K.*; Peng, K.*; Zhang, Y.*; et al.
New Journal of Physics (Internet), 25(1), p.013032_1 - 013032_11, 2023/01
Times Cited Count:0 Percentile:0.00(Physics, Multidisciplinary)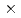 4); Comparison of X-ray diffraction and first-principles calculation
4); Comparison of X-ray diffraction and first-principles calculationTakahashi, Masamitsu; Kratzer, P.*; Penev, E.*; Mizuki, Junichiro
Surface Science, 600(18), p.4099 - 4102, 2006/09
Times Cited Count:1 Percentile:6.14(Chemistry, Physical)The GaAs(001)-c(44) has been studied by synchrotron surface X-ray diffraction. The atomic coordinates and Debye-Waller factors were determined up to the sixth layer from the surface. The results support the formation of the Ga-As heterodimers. The resultant atomic coordinates were compared with those given by a first-principle calculation. In spite of the theoretical prediction of the stability of the single-heterodimer structure, our data fit best a three-heterodimer model where three heterodimers are present in one unit cell. The preference of the formation of the three heterodimers will be discussed in the relationship with the transition process from the 24 to the c(44) structures.
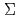 5 (012) symmetrical tilt grain boundary; A First-principles study
5 (012) symmetrical tilt grain boundary; A First-principles studyYamaguchi, Masatake; Shiga, Motoyuki; Kaburaki, Hideo
Journal of Physics; Condensed Matter, 16(23), p.3933 - 3956, 2004/06
Times Cited Count:97 Percentile:93.71(Physics, Condensed Matter)A series of non-transition elements bound to the Ni 5(012) symmetrical tilt grain boundary (GB) and the (012) free surface (FS) systems has been studied by first-principles calculation using WIEN2k code. The multilayer relaxations in presence/absence of the solutes are determined by the force minimization. The binding energies at some GB/FS/bulk sites including both interstitial and substitutional sites are calculated for all the non-transition elements between 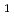 H and
H and 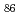 Rn. The GB/FS segregation energy is obtained by calculating the binding energy difference between the GB/FS site and the bulk site. The embrittling potency energy is obtained by calculating the difference between the GB and FS segregation energies based on Rice-Wang model. Our results show that most of the non-transition elements have negative GB/FS segregation energies. Here, this means that there exists a segregation site in the GB/FS. The embrittling potency energies are positive for most of the solutes. However, some exceptions like Be, B, C, and Si having negative and large embrittling potency can enhance the GB cohesion. Our results are found to be consistent with the experimental findings.
Rn. The GB/FS segregation energy is obtained by calculating the binding energy difference between the GB/FS site and the bulk site. The embrittling potency energy is obtained by calculating the difference between the GB and FS segregation energies based on Rice-Wang model. Our results show that most of the non-transition elements have negative GB/FS segregation energies. Here, this means that there exists a segregation site in the GB/FS. The embrittling potency energies are positive for most of the solutes. However, some exceptions like Be, B, C, and Si having negative and large embrittling potency can enhance the GB cohesion. Our results are found to be consistent with the experimental findings.
Yamaguchi, Masatake; Kaburaki, Hideo; Freeman, A. J.*
Physical Review B, 69(4), p.045408_1 - 045408_6, 2004/01
Times Cited Count:14 Percentile:55.16(Materials Science, Multidisciplinary)Multilayer relaxation at high-index Cu(hkl) (hkl = 511, 320, and 410) stepped surfaces were determined by the first-principles all-electron full-potential linearized augmented plane wave (FLAPW) method within the framework of the local density approximation (LDA) and the generalized gradient approximation (GGA). The calculated relaxation of the interlayer distances, obtained by a geometry optimization procedure that minimizes the force on each atom, were compared with low energy electron diffraction (LEED) analysis of experimental data. In the case of Cu(511), the calculated results are in good agreement with the LEED analyses. On the other hand, for Cu(320) and Cu(410), there are large differences that may be understood from the fact that the LEED analyses of experiments consider up to only three or four layers from the surface, and that whereas even the fifth or sixth layers show large relaxation in our calculations. Our results suggest a reanalysis of the LEED data with the inclusion of more layers.
; Chihara, Junzo
Physical Review E, 53(6), p.6253 - 6263, 1996/06
Times Cited Count:16 Percentile:57.82(Physics, Fluids & Plasmas)no abstracts in English
